A new MIT study from the Nedivi Lab finds that somatostatin-expressing neurons follow a unique trajectory when forming connections in the brain’s visual cortex that may help establish the conditions needed for sensory experience to refine circuits.
David Orenstein | The Picower Institute for Learning and Memory
January 14, 2026
The way the brain develops can shape us throughout our lives, so neuroscientists are intensely curious about how it happens. A new study by researchers in The Picower Institute for Learning and Memory at MIT that focused on visual cortex development in mice, reveals that an important class of neurons follows a set of rules that while surprising, might just create the right conditions for circuit optimization.
During early brain development, multiple types of neurons emerge in the visual cortex (where the brain processes vision). Many are “excitatory,” driving the activity of brain circuits, and others are “inhibitory,” meaning they control that activity. Just like a car needs not only an engine and a gas pedal, but also a steering wheel and brakes, a healthy balance between excitation and inhibition is required for proper brain function. During a “critical period” of development in the visual cortex, soon after the eyes first open, excitatory and inhibitory neurons forge and edit millions of connections, or synapses, to adapt nascent circuits to the incoming flood of visual experience. Over many days, in other words, the brain optimizes its attunement to the world.
In the new study in The Journal of Neuroscience, a team led by MIT research scientist Josiah Boivin and Professor Elly Nedivi visually tracked somatostatin (SST)-expressing inhibitory neurons forging synapses with excitatory cells along their sprawling dendrite branches, illustrating the action before, during and after the critical period with unprecedented resolution. Several of the rules the SST cells appeared to follow were unexpected—for instance, unlike other cell types, their activity did not depend on visual input—but now that the scientists know these neurons’ unique trajectory, they have a new idea about how it may enable sensory activity to influence development: SST cells might help usher in the critical period by establishing the baseline level of inhibition needed to ensure that only certain types of sensory input will trigger circuit refinement.
“Why would you need part of the circuit that’s not really sensitive to experience? It could be that it’s setting things up for the experience-dependent components to do their thing,” said Nedivi, William R. and Linda R. Young Professor in The Picower Institute and MIT’s Departments of Biology and Brain and Cognitive Sciences.
Boivin added: “We don’t yet know whether SST neurons play a causal role in the opening of the critical period, but they are certainly in the right place at the right time to sculpt cortical circuitry at a crucial developmental stage.”
A unique trajectory
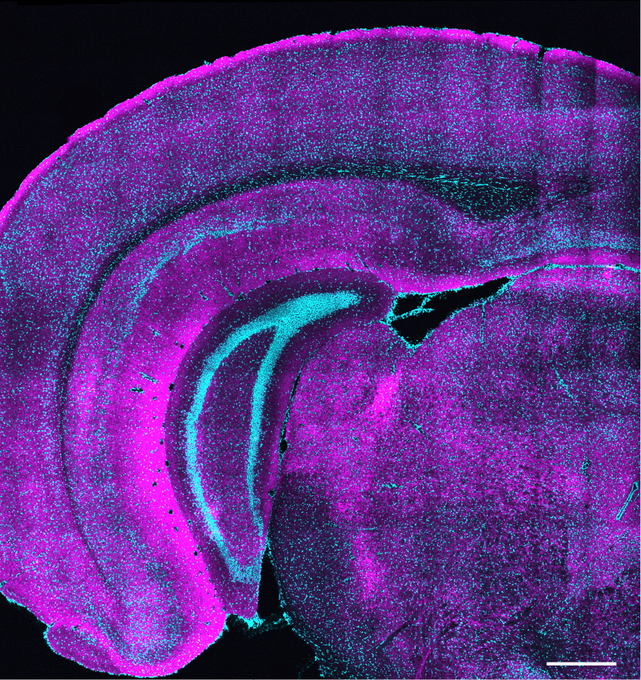
To visualize SST-to-excitatory synapse development, Nedivi and Boivin’s team used a genetic technique that pairs expression of synaptic proteins with fluorescent molecules to resolve the appearance of the “boutons” SST cells use to reach out to excitatory neurons. They then performed a technique called eMAP, developed by Kwanghun Chung’s lab in the Picower Institute, that expands and clears brain tissue to increase magnification, allowing super-resolution visualization of the actual synapses those boutons ultimately formed with excitatory cells along their dendrites. Co-author and postdoc Bettina Schmerl helped lead the eMAP work.
These new techniques revealed that SST bouton appearance and then synapse formation surged dramatically when the eyes opened and then as the critical period got underway. But while excitatory neurons during this timeframe are still maturing, first in the deepest layers of the cortex and later in its more superficial layers, the SST boutons blanketed all layers simultaneously, meaning that, perhaps counter intuitively, they sought to establish their inhibitory influence regardless of the maturation stage of their intended partners.
Many studies have shown that eye opening and the onset of visual experience sets in motion the development and elaboration of excitatory cells and another major inhibitory neuron type (parvalbumin-expressing cells). Raising mice in the dark for different lengths of time, for instance, can distinctly alter what happens with these cells. Not so for the SST neurons. The new study showed that varying lengths of darkness had no effect on the trajectory of SST bouton and synapse appearance; it remained invariant, suggesting it is pre-ordained by a genetic program or an age-related molecular signal, rather than experience.
Moreover, after the initial frenzy of synapse formation during development, many synapses are then edited, or pruned away, so that only the ones needed for appropriate sensory responses endure. Again, the SST boutons and synapses proved to be exempt from these redactions. Though the pace of new SST synapse formation slowed at the peak of the critical period, the net number of synapses never declined and even continued increasing into adulthood.
“While a lot of people think that the only difference between inhibition and excitation is their valence, this demonstrates that inhibition works by a totally different set of rules,” Nedivi said.
In all, while other cell types were tailoring their synaptic populations to incoming experience, the SST neurons appeared to provide an early but steady inhibitory influence across all layers of the cortex. After excitatory synapses have been pruned back by the time of adulthood, the continued upward trickle of SST inhibition may contribute to the increase in the inhibition to excitation ratio that still allows the adult brain to learn, but not as dramatically or as flexibly as during early childhood.
A platform for future studies
In addition to shedding light on typical brain development, Nedivi said, the study’s techniques can enable side-by-side comparisons in mouse models of neurodevelopmental disorders such as autism or epilepsy where aberrations of excitation and inhibition balance are implicated.
Future studies using the techniques can also look at how different cell types connect with each other in brain regions other than the visual cortex, she added.
Boivin, who will soon open his own lab as a faculty member at Amherst College, said he is eager to apply the work in new ways.
“I’m excited to continue investigating inhibitory synapse formation on genetically defined cell types in my future lab,” Boivin said. “I plan to focus on the development of limbic brain regions that regulate behaviors relevant to adolescent mental health.”
In addition to Nedivi, Boivin and Schmerl, the paper’s other authors are Kendyll Martin, and Chia-Fang Lee.
Funding for the study came from the National Institutes of Health, the Office of Naval Research and the Freedom Together Foundation.