Cell biologist Whitney Henry and immunologist Harikesh Wong will receive four years of flexible funding to advance early-career research on ferroptosis and immune decision-making.
Nina Tamburello | Nikolay Kolev | Koch Institute | Ragon Institute
June 29, 2026
Whitney Henry and Harikesh Wong have been named 2026 Pew Scholars in the Biomedical Sciences. The Pew Charitable Trusts announced the 21-member class of early-career researchers, which includes the two MIT scientists as well as two alumni, on June 16. Each scholar will receive four years of funding to pursue cutting-edge research into human health and disease. Xin Gu PhD ’22 of Dana-Farber Cancer Institute and Christina Tringides ’15 of Rice University were also selected as scholars.
Henry, the Robert A. Swanson (1969) Career Development Professor of Life Sciences and a faculty member at the Koch Institute for Integrative Cancer Research, will use the Pew scholarship to examine how a stress-induced cell death program called ferroptosis contributes to injury and regeneration in the liver. Wong, assistant professor of biology at MIT and core member at the Ragon Institute of Mass General Brigham, MIT, and Harvard, will use his award to investigate how groups of immune cells reach a “communal decision” about whether to tolerate or attack a particular target.
Whitney Henry
Henry’s research centers on ferroptosis — an iron-dependent form of regulated cell death — and its role in shaping cell fate and tissue remodeling. Her lab investigates why some cells can withstand stress while others cross the threshold for ferroptosis, focusing on the molecular, metabolic, and tissue-level cues that shape ferroptosis vulnerability. The work draws on chemical biology, metabolomics, functional genomics, and in vivo models. By defining the mechanisms that govern ferroptosis susceptibility, Henry’s group aims not only to identify novel therapies that target the most dangerous subpopulations of cancer cells, those that are highly metastatic and resistant to conventional treatment, but also to advance understanding of diseases in which ferroptosis drives tissue injury, fibrosis, or impaired repair.
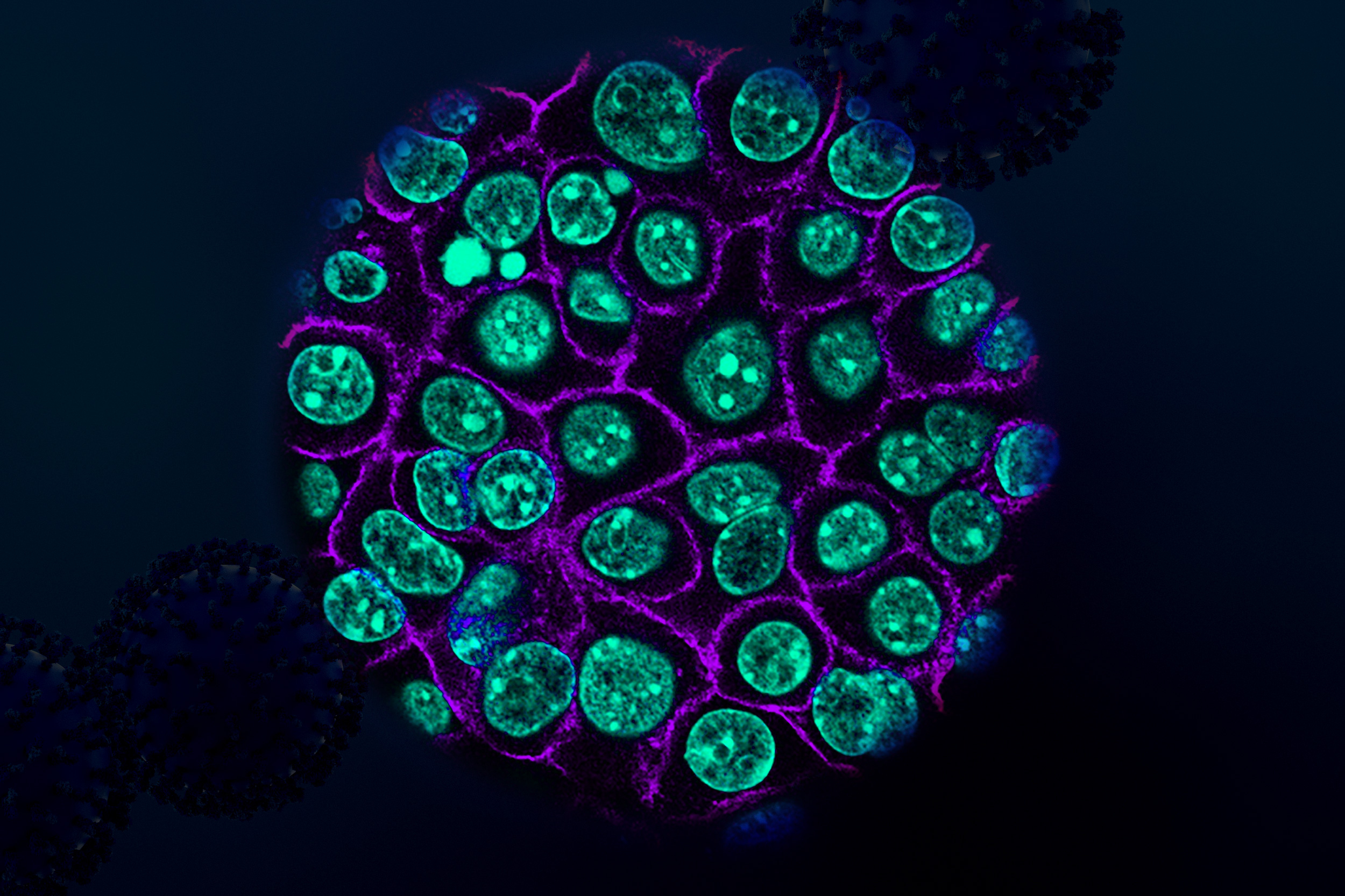
Harikesh Wong
Wong investigates how groups of cells organize into networks that collectively process information and control immune responses within tissues. These networks must continually balance the body’s need to protect itself against pathogens and tumors with the need to preserve healthy tissue function. Combining the tools of immunology with high-resolution fluorescence microscopy, computational modeling, and gene manipulation, his lab seeks to map, model, and manipulate the cell-cell interactions that govern these decisions within intact tissues, revealing how subtle changes in multicellular organization and communication can shift immune responses toward pathogen clearance and tolerance, or toward autoimmunity, chronic inflammation, and cancer.
Pew scholars are chosen from applicants nominated by leading academic institutions across the United States. This year’s class of 21 was selected from 211 nominees. The incoming scholars join a legacy of more than 1,000 scientists supported by the program since 1985. During their time as scholars, they will meet annually with fellow Pew-funded scientists to build connections across a wide variety of disciplines.
“Scientific discovery is moving at a rapid pace, and now more than ever we need curious and creative researchers leading the charge,” says Lee Niswander, a 1995 Pew scholar and chair of the program’s national advisory committee. “These new biomedical scholars are prepared to meet that challenge, and I look forward to watching their research unfold.”