SNIPE, a newly characterized defense system, directly protects bacteria by chopping up invading viral DNA.
Lillian Eden | Department of Biology
April 9, 2026
What if the Trojan horse had been pulled to pieces, revealing the ruse and fending off the invasion, just as it entered the gates of Troy?
That’s an apt description of a newly characterized bacterial defense system that chops up foreign DNA.
Bacteria and the viruses that infect them, bacteriophages — phages for short — are ceaselessly at odds, with bacteria developing methods to protect themselves against phages that are constantly striving to overcome those safeguards.
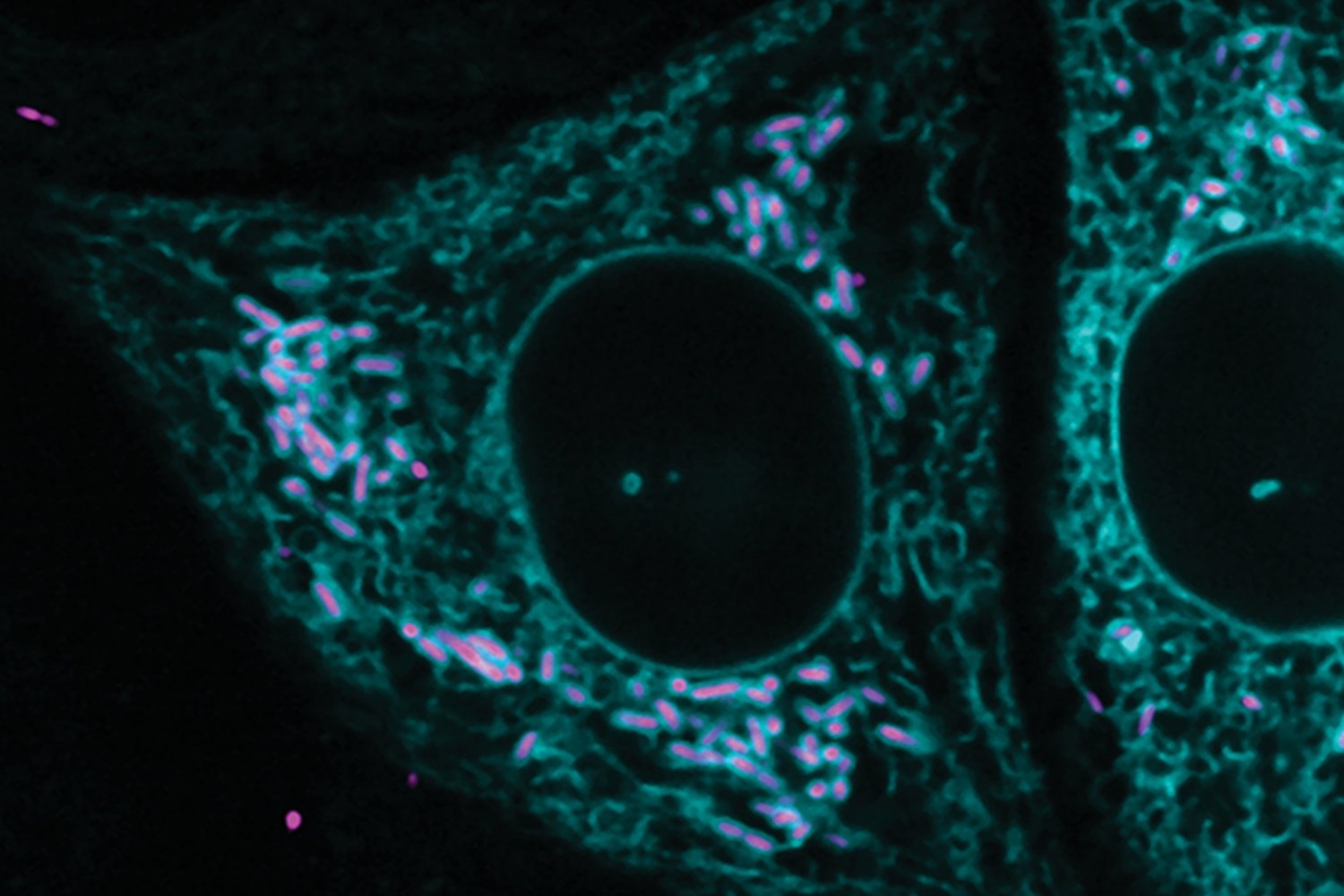
New research from the Department of Biology at MIT, recently published in Nature, describes a defense system that is integrated into the protective membrane that encapsulates bacteria. SNIPE, which stands for surface-associated nuclease inhibiting phage entry, contains a nuclease domain that cleaves genetic material, chopping the invading phage genome into harmless fragments before it can appropriate the host’s molecular machinery to make more phages.
Daniel Saxton, a postdoc in the Laub Lab and the paper’s first author, was initially drawn to studying this bacterial defense system in E. coli, in part because it is highly unusual to have a nuclease that localizes to the membrane, as most nucleases are free-floating in the cytoplasm, the gelatinous fluid that fills the space inside cells.
“The other thing that caught my attention is that this is something we call a direct defense system, meaning that when a phage infects a cell, that cell will actually survive the attack,” Saxton says. “It’s hard to fend off a phage directly in a cell and survive — but this defense system can do it.”
Light it up
For Saxton, the project came into focus during a fluorescence-based experiment in which viral genetic material would light up if it successfully penetrated the bacteria.
“SNIPE was obliterating the phage DNA so fast that we couldn’t even see a fluorescent spot,” Saxton recalls. “I don’t think I’ve ever seen such an effective defense system before — you can barrage the bacteria with hundreds of phage per cell, but SNIPE is like god-tier protection.”
When the nuclease domain of SNIPE was mutated so it couldn’t chop up DNA, fluorescent spots appeared as usual, and the bacteria succumbed to the phage infection.
Bacteria maintain tight control over all their defense systems, lest they be turned against their host. Some systems remain dormant until they flare up, for example, to halt all translation of all proteins in the cell, while others can distinguish between bacterial DNA and foreign, invading phage DNA. There were only two previously characterized mechanisms in the latter category before researchers uncovered SNIPE.
“Right now, the phage field is at a really interesting spot where people are discovering phage defense systems at a breakneck pace,” Saxton says.
Problems at the periphery
Saxton says they had to approach the work in a somewhat roundabout way because there are currently no published structures depicting all the steps of phage genome injection. Studying processes at the membrane is challenging: Membranes are dense and chaotic, and phage genome injection is a highly transient process, lasting only a few minutes.
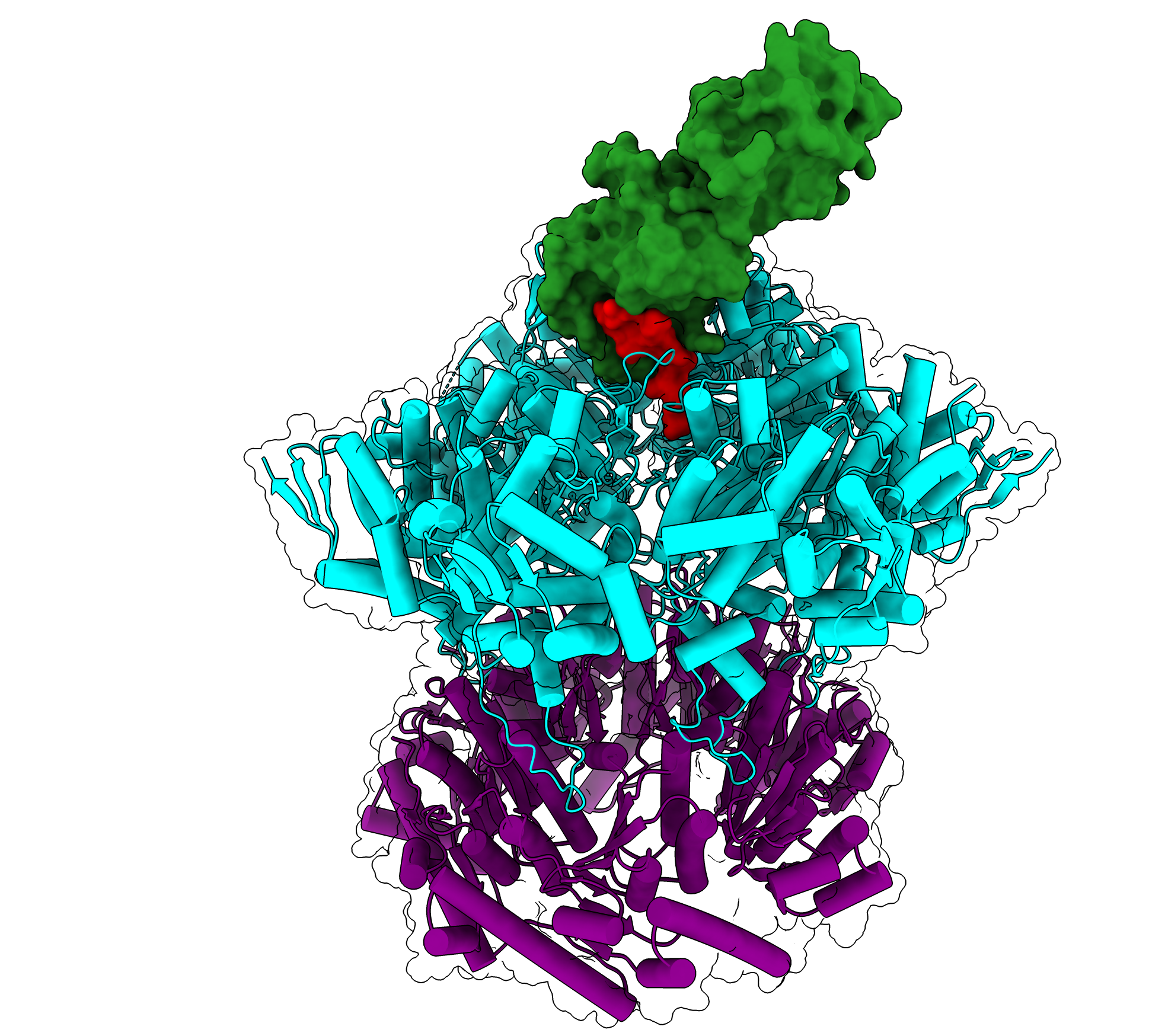
SNIPE seems to discern viral DNA by interacting with proteins the phage uses to tunnel through the bacteria’s protective membrane. This “subcellular localization,” according to Saxton, may also prevent SNIPE from inadvertently chopping up the bacteria’s own genetic material.
The model outlined in the paper is that one region of SNIPE binds to a bacterial membrane protein called ManYZ, while another region likely binds to the tape measure protein from the phage.
The tape measure protein got its name because it determines the length of the phage tail — the part of the phage between the small, leglike protrusions and the bulbous head, which contains the phage’s genetic material. The researchers revealed that the phage’s tape measure protein enters the cytoplasm during injection, a phenomenon that had not been physically demonstrated before.
There may also be other proteins or interactions involved.
“If you shunt the phage genome injection through an alternate pathway that isn’t ManYZ, suddenly SNIPE doesn’t defend against the phage nearly as well,” Saxton says. “It’s unclear exactly how these proteins interact, but we do know that these two proteins are involved in this genome injection process.”
Future directions
Saxton hopes that future work will expand our understanding of what occurs during phage genome injection and uncover the structures of the proteins involved, especially the tunnel complex in the membrane through which phages insert their genome.
Members of the Laub Lab are already collaborating with another lab to determine the structure of SNIPE. In the meantime, Saxton has been working on a new defense system in which molecular mimicry — bacterial proteins imitating phage proteins — may play a role.
Michael T. Laub, the Salvador E. Luria Professor of Biology and a Howard Hughes Medical Institute investigator, notes that one of the breakthrough experiments for demonstrating how SNIPE works came from a brainstorming session at a lab retreat.
“Daniel and I were kind of stuck with how to directly measure the effect of SNIPE during infection, but another postdoc in the lab, Ian Roney, who is a co-author on the paper, came up with a very clever idea that ultimately worked perfectly,” Laub recalls. “It’s a great example of how powerful internal collaborations can be in pushing our science forward.”