Newly developed model-based analysis reveals protein proofreading in ribosome biogenesis
Research from the Davis Lab in the Department of Biology at MIT, in collaboration with the Ortega Group at McGill University, showcases the power of machine learning to solve biological puzzles
By Lillian Eden
One of the most critical complexes in cells is the ribosome, the large and elaborate machinery responsible for translating genetic code into proteins–but how do cells make and assemble the ribosomes?
Assembly of ribosomes, which are formed from a delicately interwoven mesh of proteins and RNA, requires the help of many proteins that are generally referred to as assembly factors. Assembly factors perform a range of functions, including catalyzing ribosomal RNA folding or promoting ribosomal protein binding. These factors are especially important in stressful conditions, such as cold temperatures, where ribosomal RNA can become kinetically trapped in misfolded states.
Because of the complex multi-step nature of ribosome assembly and the sheer number of assembly factors involved, the role of many assembly factors has yet to be defined.
Of the assembly factors involved in prokaryotic ribosome biogenesis, more than 20 are from a class of enzymes called methyltransferases that transfer a methyl group to ribosomal RNA. The contributions of these methyltransferases to ribosome assembly remain unclear.
Research recently published in Nature Structural & Molecular Biology from the Davis Lab in the Department of Biology at MIT reveals that a methyltransferase called KsgA provides quality control proofreading for ribosome assembly.
Although scientists have theorized biological proofreading for decades, to the authors’ knowledge, this is the first time an assembly factor has been shown to have a proofreading role in ribosome biogenesis.
The work, performed in collaboration with researchers in the Ortega Group in the Department of Anatomy and Cell Biology at McGill University, showcases the power of machine learning to solve biological puzzles. The paper outlines an innovative approach to structural analysis that allowed the researchers to make quantitative comparisons of structural differences between two different experimental conditions—isolated ribosomal small subunits treated with KsgA compared to untreated subunits.
In the presence of KsgA, fully assembled subunits were flawless. Subunits not treated with KsgA appeared fully assembled. However, closer inspection revealed defects—key components were not in the correct positions. KsgA binding seems to induce a cascade of changes that caused the subunit to partially disassemble, giving the subunit another chance to assemble correctly.
Co-first author Laurel Kinman, a graduate student in the Davis lab, was able to parse out structural differences in a systematic and quantifiable way using cryo-electron microscopy, or cryo-EM and a novel, high throughput approach, which the researchers termed Model-based Analysis of Volume Ensembles, or MAVEn for short. Cryo-EM is a powerful tool that involves freezing samples in a thin layer of ice and imaging them; researchers use a composite of thousands of 2D images to generate 3D structures.
By their very nature, however, proteins are dynamic, constantly changing shape. According to Kinman, it is interesting and valuable to measure these changes in structure and infer how structure determines function. But it is also challenging to capture those differences when structures are generated through averaging—a structural variation that only occurs in a small fraction of the particles is likely to be lost in the noise of a more common conformation.
To address this problem, the Davis Lab previously developed cryoDRGN, a neural network-based machine learning program that uses 2D images to generate a wide array of 3D structures. Traditional approaches rely on iterative rounds of expert-guided classification, often producing only a small number of 3D structures. By contrast, cryoDRGN’s machine-learning approach allows researchers to generate hundreds to thousands of maps from a single dataset. Despite the power of the cryoDRGN approach, however, strategically analyzing and interpreting so many volumes remains challenging and labor-intensive.
To identify and measure key structural differences between these volumes, the researchers developed the MAVEn approach outlined in the paper. This approach took advantage of the fact that much is understood about where ribosomal components should be located when the ribosome is assembled properly by measuring how much volume was present in a given subunit’s expected location. Doing so across many dozens of subunits allows the researchers to determine a complete picture of the particle’s structural state.
The researchers applied this approach to a dataset of ribosomal particles assembled in the absence of KsgA and a dataset where KsgA was added to purified ribosomal particles in vitro. By comparing these results, the researchers could identify and compare the frequency of structural states between the two conditions. According to Kinman, this quantitative comparison is not typically how structural biologists approach this type of data.
“One of the reasons we’re excited about this is that we think this is a really nice case study for a pipeline that can capture rare, potentially biologically informative states, and we’re able to validate our results using traditional tools,” Kinman says. “There’s a possibility that people are under-exploring their heterogenous cryo-EM data.”
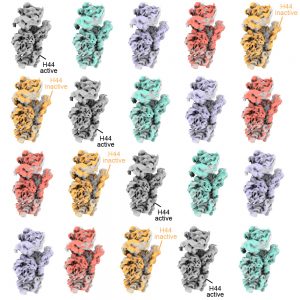
In particular, in the untreated subunits, MAVEn revealed that a component called Helix 44 often occupied an incorrect position—not fully occupying the space it should. This component is critical because if Helix 44 is incorrectly positioned in a fully assembled ribosome, the ribosome cannot proceed with translation. By contrast, in subunits treated with KsgA, there were no mature subunits with inactive Helix 44; however, many more of these particles also lacked other features, indicating they were not fully assembled. Taken together, these findings suggested that KsgA was involved in selectively pruning the inactive conformation of Helix 44 by inducing large-scale disassembly and reassembly. This may effectively perform quality control on the assembled ribosomes to ensure they have matured correctly.
Because the subunits treated with KsgA were captured in so many varied states of assembly, Kinman was also able to use this approach to explore structural interdependencies between different elements of the ribosome. She used this data to determine how certain elements of the ribosome depended on others to assemble.
Kinman says she is excited to apply this approach to other heterogenous data sets; she’s also working on related software for when researchers don’t have an atomic model to reference or for researchers interested in visualizing conformational changes of moving parts.
Senior author Joey Davis says they’re also interested in discovering more about ribosome biogenesis.
“I think the field, generally, is trying to figure out how what we learn in these purified samples relates to what’s going on inside the cell,” Davis says. “The hope is that the combination of this paper and related work in my group will allow us to look at ribosome biogenesis in living cells.”