In certain species of bacteria, the answer lies in shielding RNA transcripts from a quality-control factor called Rho. Understanding the requirements for expressible sequences is critical for expression engineering of therapeutic agents.
Lillian Eden | Department of Biology
July 2, 2026
In the study of bacteria, a longstanding dogma held that two molecular machines — RNA polymerase, which leads the way in transcribing DNA into RNA, and ribosomes, which bring up the rear translating RNA into proteins — worked so closely in tandem that they were effectively attached.
This close coupling of transcription and translation in bacteria was thought to be fundamental to gene expression in part because the trailing ribosome could shield nascent gene products from an effective and omnipresent quality-control protein called Rho.
In bacteria that exhibit something called runaway transcription, however, the polymerase instead speeds ahead, unhitched from its protective ribosome. Inexplicably, however, in bacteria that exhibit this runaway transcription, such as Bacillus subtilis, Rho targeted primarily noncoding, useless RNA products.
New research from the Department of Biology reveals that the secret to Rho’s quality-control specificity lies in the sequence composition of nucleotide bases that make up coding strands of DNA.
“We started with a hypothesis that Rho was regulated by sequence, but the fact that the sequence alone was enough to protect any gene in the entire B. subtilis genome from Rho was really surprising,” says Julia Dierksheide PhD ’26, a graduate student in the Li Lab and first author of a paper recently published in Nature Microbiology. “That’s a really diverse range of sequences — what sequence feature is shared by every single gene in the genome?”
Barricading with bias
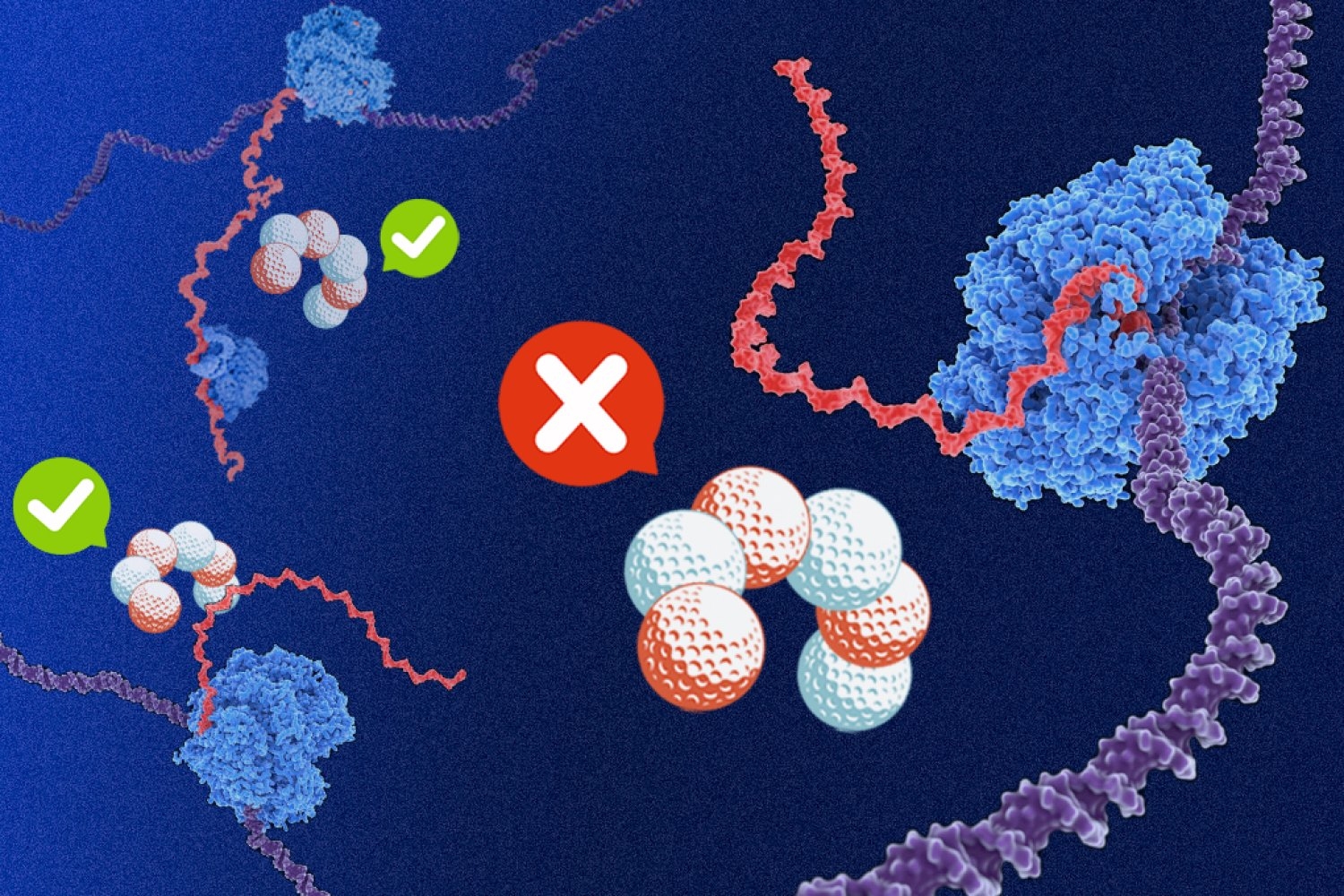
Rho serves as a termination factor, meaning that it is a crucial mechanism for preventing bacteria from wasting precious resources by making RNA transcripts that serve no purpose.
All the information a bacterial cell needs is encoded in its DNA, which is made up of two strands of nucleic acids. These strands twist together to form a double helix, with genetic information codified in pairs of bases: purines guanine and adenine are matched with pyrimidines cytosine and thymine, respectively. Any sequence that gives rise to RNA transcripts is stored in complement to a parallel, noncoding strand, meaning that a large portion of genetic material is transcriptionally useless.
Coding DNA strands in certain bacteria were known to be significantly higher in purines guanine and adenine compared to the rest of the bacterial genome. The researchers found that this purine bias alone shields productive mRNA transcripts from Rho-mediated termination.
“I love having a big, complicated dataset and trying to reduce that to biological meaning,” Dierksheide says. “It seems like Rho itself has been broadly shaping the evolution of the B. subtilis genome to create these sequence composition biases.”
Bacterial species that, over generations, have lost Rho no longer exhibit this strong purine bias.
Rho also serves as a regulatory factor in bacteria becoming motile, forming biofilms, or sporulating, all of which are critical for biology and survival. The purine bias could also provide a layer of protection against the insertion of foreign DNA, for example, when a viral bacteriophage infects bacteria.
“Bacteria exist as single cells, so everything that they do, they have to do through gene expression,” Dierksheide says. “Understanding the fundamental details about how gene expression works, how a cell encodes all the information it needs to survive in the nucleotide sequence of the genome, is really exciting.”
Future directions
Although the exact mechanism underlying Rho’s specificity remains unclear, these results crack an underlying code in the composition of bacterial genomes.
Dierksheide said she hoped to perform a similar screen to characterize Rho’s specificity in Escherichia coli, which diverged from B. subtilis on the evolutionary tree an estimated 2 billion years ago and still exhibits coupled transcription-translation, where the transcribing RNA polymerase is closely followed by a translating ribosome.
The high sequence specificity of B. subtilis Rho is crucial for the protection of its runaway RNA polymerase, in which that molecular machine speeds ahead of the ribosome. A systematic comparison to E. coli Rho could help reveal how this heightened stringency arose.
This information will be critical for engineering diverse bacterial species for applications including the production of therapeutic agents. Other bacterial species, such as B. subtilis, may be better models for this process because they have abundant secretion pathways, according to Dierksheide, making it much easier to produce and isolate proteins in large quantities.
“Our findings reveal an important criterion for successful sequence design that must be considered in expression engineering,” says associate department head, associate professor of biology, and Howard Hughes Medical Institute investigator Gene-Wei Li, the lead author of the study. “There are so many cryptic messages in the genome, like the purine bias, and we are just beginning to be able to decipher what they mean.”