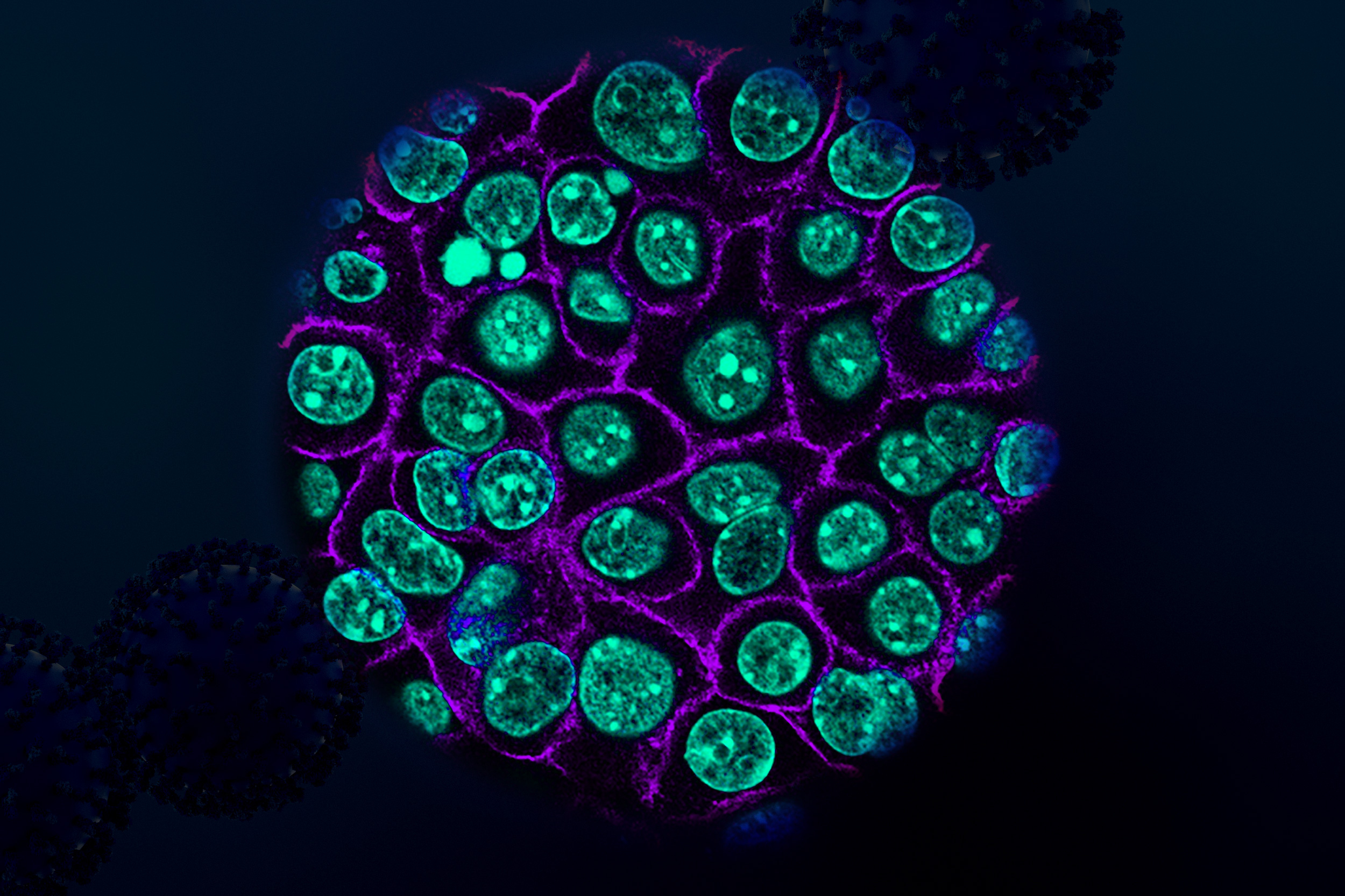
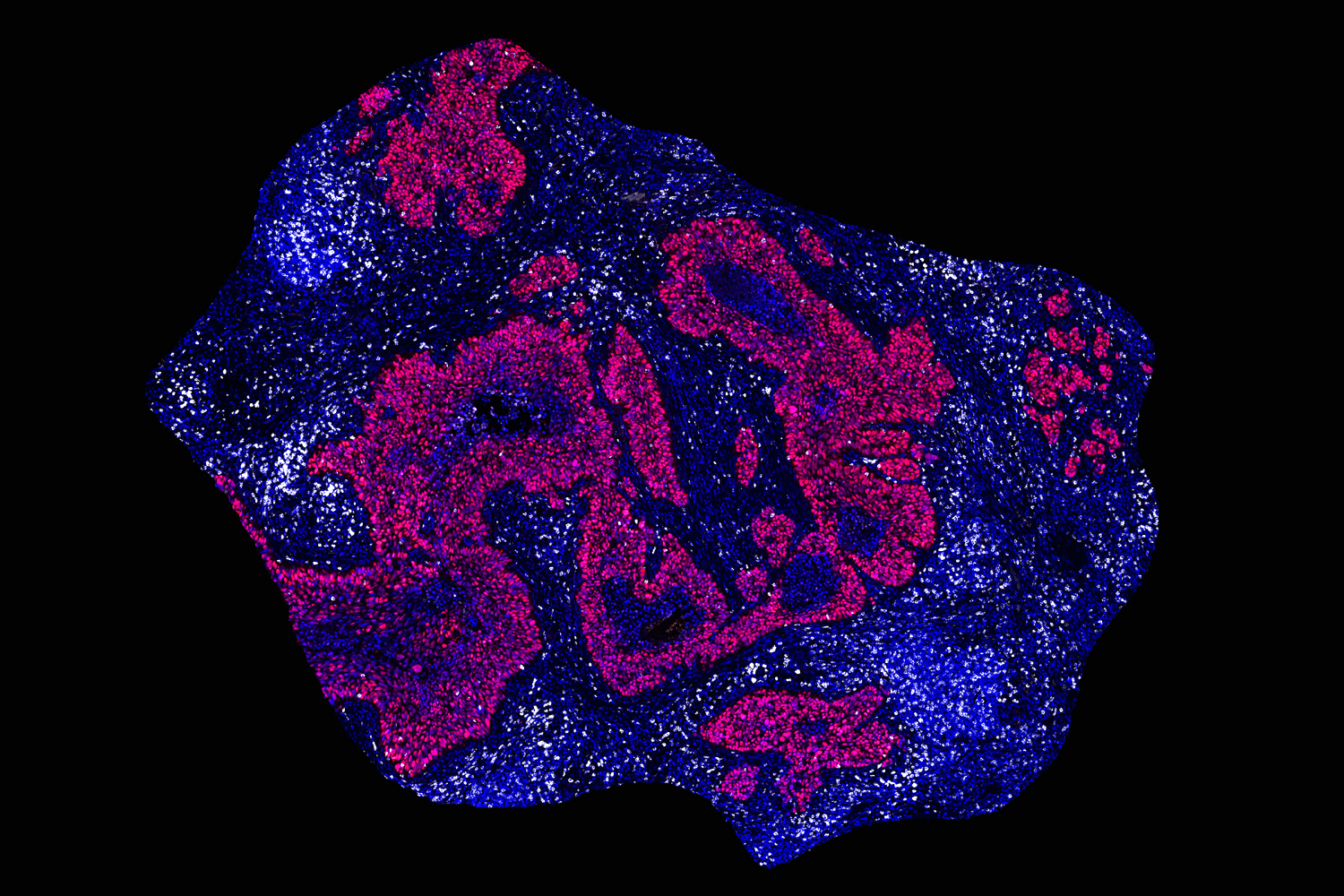
The assistant professor hopes to decode molecular processes on the genetic, epigenetic, and microenvironment levels to anticipate how and when tumors evolve to resist treatment.
Lillian Eden | Department of Biology
March 10, 2026
Just as Darwin’s finches evolved in response to natural selection in order to endure, the cells that make up a cancerous tumor similarly counter selective pressures in order to survive, evolve, and spread. Tumors are, in fact, complex sets of cells with their own unique structure and ability to change.
Today, artificial Intelligence and machine learning tools offer an unparalleled opportunity to illuminate the generalizable rules governing tumor progression on the genetic, epigenetic, metabolic, and microenvironmental levels.
Matthew G. Jones, an Assistant Professor in the Department of Biology at MIT, the Koch Institute for Integrative Cancer Research, and the Institute for Medical Engineering and Science, hopes to use computational approaches to build predictive models — to play a game of chess with cancer, making sense of a tumor’s ability to evolve and resist treatment with the ultimate goal of improving patient outcomes.
Q: What aspect of tumor progression are you hoping to explore and characterize?
A: A very common story with cancer is that patients will respond to a therapy at first, and then eventually that treatment will stop working. The reason this largely happens is that tumors have an incredible, and very challenging, ability to evolve: the ability to change their genetic makeup, protein signaling composition, and cellular dynamics. The tumor as a system also evolves at a structural level. Oftentimes, the reason why a patient succumbs to a tumor is because either the tumor has evolved to a state we can no longer control, or it evolves in an unpredictable manner.
In many ways, cancers can be thought of as, on the one hand, incredibly dysregulated and disorganized, and on the other hand, as having their own internal logic, which is constantly changing. The central thesis of my lab is that tumors follow stereotypical patterns in space and time, and we’re hoping to use computation and experimental technology to decode the molecular processes underlying these transformations.
We’re focused on one specific way tumors are evolving through a form of DNA amplification called extrachromosomal DNA. Excised from the chromosome, these ecDNAs are circularized and exist as their own separate pool of DNA particles in the nucleus.
Initially discovered in the 1960s, ecDNA were thought to be a rare event in cancer. However, as researchers began applying next-generation sequencing to large patient cohorts in the 2010s, it seemed like not only were these ecDNA amplifications conferring the ability of tumors to adapt to stresses, and therapies, faster, but that they were far more prevalent than initially thought.
We now know these ecDNA amplifications are apparent in about 25% of cancers, in the most aggressive cancers: brain, lung, and ovarian cancers. We have found that, for a variety of reasons, ecDNA amplifications are able to change the rule book by which tumors evolve in ways that allow them to accelerate to a more aggressive disease in very surprising ways.
Q: How are you planning to use machine learning and artificial intelligence to study ecDNA amplifications and tumor evolution?
A: There’s a mandate to translate what I’m doing in the lab to improve patients’ lives. I want to start with patient data to discover how various evolutionary pressures are driving disease and the mutations we observe.
One of the tools we use to study tumor evolution is single-cell lineage tracing technologies. Broadly, they allow us to study the lineages of individual cells. When we sample a particular cell, not only do we know what that cell looks like, but we can, ideally, pinpoint exactly when aggressive mutations appeared in the tumor’s history. That evolutionary history gives us a way of studying these dynamic processes that we otherwise wouldn’t be able to observe in real time and helps us make sense of how we might be able to intercept that evolution.
I hope we’re going to get better at stratifying patients who will respond to certain drugs, to anticipate and overcome drug resistance, and to identify new therapeutic targets.
Q: What excites you about joining this community, and what sorts of trainees are you hoping to recruit to your lab?
A: One of the things that I was really attracted to was the integration of excellence in both engineering and biological sciences. At the Koch Institute, every floor is structured to promote this interface between engineers and basic scientists, and beyond campus, we can connect with all the biomedical research enterprises in the Greater Boston Area.
Another thing that drew me to MIT was the fact that it places such a strong emphasis on education, training, and investing in student success. I’m a personal believer that what distinguishes academic research from industry research is that academic research is fundamentally a service job, in that we are training the next generation of scientists.
It was always a mission of mine to bring excellence to both computational and experimental technology disciplines. The types of trainees I’m hoping to recruit are those who are eager to collaborate and solve big problems that require both disciplines. The KI is uniquely set up for this type of hybrid lab: my dry lab is right next to my wet lab, and it’s a source of collaboration and connection, and that reflects the KI’s general vision.