




In late February, Vice Chancellor for Undergraduate and Graduate Education Ian A. Waitz and Faculty Chair Mary Fuller announced the formation and launch of the Task Force on the MIT Undergraduate Academic Program (TFUAP). The effort fulfills a critical recommendation of the Task Force 2021 and Beyond RIC1 (Undergraduate Program) and draws upon several, prior foundational working groups — some focused on the current General Institute Requirements (GIRs) and others on updating recent studies for the purposes of this review.
In this interview, task force co-chairs Adam Martin, professor of biology, and Joel Voldman, the William R. Brody Professor of Electrical Engineering and Computer Science describe the TFUAP’s goals, approach, and next steps.
Q: The charge of the task force is quite ambitious, including “reviewing the current undergraduate academic program and considering improvements with a focus on both the curriculum and pedagogy.” Can you explain your approach?
Martin: For context, it’s important to know that the undergraduate program is multifaceted and consists of many components, including majors, electives, experiential learning, and of course the GIRs — arguably one of the best-known acronyms at MIT! Moreover, the GIRs include science core classes; humanities, arts, and social sciences classes; certain electives in science and engineering; and a lab requirement, each of which serves a slightly different purpose and dovetails with majors and minors in unique ways.
Some aspects of the academic program are determined by the faculty, either MIT-wide or within a particular department. Others can be customized by students, in consultation with faculty and staff advisors, from the broad array of curricular and co-curricular offerings at MIT. The task force will look holistically at all of these aspects, considering both what MIT requires of all students, and the options we make available as students chart their own paths.
As part of this holistic approach, the TFUAP will zero in on both content and pedagogy. Obviously, the content we cover is important; our goal must remain to provide undergraduates with the world-class education they expect. But how we teach is of fundamental importance, as well. The pedagogy we adopt should be inclusive, supported by research, and designed to help students not only understand what they are learning, but why they are learning it — how it relates to their majors, potential careers, and their lives.
Voldman: I think your question’s description of our charge as “ambitious” is noteworthy. We feel that the task force is ambitious, too, but perhaps in a different sense from the question. That is, we believe our job is to not only think about nuts-and-bolts issues of the academic program requirements but also to consider the big picture. What are the most expansive possibilities? How can we push the envelope? That’s the MIT way, after all.
Q: The task force is building upon quite a bit of past work and benefits from some major accomplishments recommended by Task Force 2021 (TF2021). For example, how does the creation of the Undergraduate Advising Center, and in general, the desire to provide more personal and professional support to all students, fit in with the potential updates to the undergraduate curriculum?
Martin: You’re absolutely right — our work benefits greatly from years of conversations focused on the undergraduate academic program, particularly in the last decade or so. These include the 2014 Task Force on the Future of Education; the 2018 Designing the First-Year Experience Class; Task Force 2021 and Beyond (TF2021); the Foundational Working Groups (part of the RIC 1 implementation) that have studied the existing MIT undergraduate program; and the Committee on the Undergraduate Program. The valuable work of these past committees and their findings will certainly inform our thought process.
In the past, groups that evaluated the undergraduate curriculum were also charged with tackling related topics, such as undergraduate advising or revamping classrooms. Taking on any one of these three issues is ambitious by any measure! What’s changed in the past decade is that advances have been made in these other critical areas, so the TFUAP can focus solely on curriculum and pedagogy. For example, thanks to recent accomplishments by TF2021 and others, we have implemented a new advising system for all undergraduates in the form of the Undergraduate Advising Center.
We envision the TFUAP being a highly collaborative process, bringing in voices across the entire Institute and beyond. We welcome input from members of the community via email at tfuap@mit.edu. We will also be reaching out to student groups, alumni, individual faculty, faculty groups, and administrative staff across the Institute to hear their perspectives.
Q: Part of what TFUAP will have to confront, no doubt, are some of the most pressing issues of our time, like the rise of computing and AI, climate change (what President Kornbluth calls an existential threat to our way of life), and the changing nature of learning (online, hybrid, etc.). How are you thinking about all of these factors?
Voldman: That is a good question! It’s early days, and our work is just beginning, but we know that these and other issues loom over all of us. For example, we are keenly aware of the influx of students into computing-related majors and classes, and we need to think deeply about the implications. Furthermore, we want a curriculum that prepares students for current and upcoming global challenges as well as changes in the technology and tools available to address those challenges. However, we can expect that our students will need to be agile and curious, lifelong learners, collaborative and compassionate teammates, and creative and thoughtful problem-solvers.
As we work with the community to design the next version of an MIT undergraduate education, it will be important to build a structure that can incorporate the biggest challenges and opportunities of the day, while staying flexible and responsive to an ever-evolving world.


Tumors can carry mutations in hundreds of different genes, and each of those genes may be mutated in different ways — some mutations simply replace one DNA nucleotide with another, while others insert or delete larger sections of DNA.
Until now, there has been no way to quickly and easily screen each of those mutations in their natural setting to see what role they may play in the development, progression, and treatment response of a tumor. Using a variant of CRISPR genome-editing known as prime editing, MIT researchers have now come up with a way to screen those mutations much more easily.
The researchers demonstrated their technique by screening cells with more than 1,000 different mutations of the tumor suppressor gene p53, all of which have been seen in cancer patients. This method, which is easier and faster than any existing approach, and edits the genome rather than introducing an artificial version of the mutant gene, revealed that some p53 mutations are more harmful than previously thought.
This technique could also be applied to many other cancer genes, the researchers say, and could eventually be used for precision medicine, to determine how an individual patient’s tumor will respond to a particular treatment.
“In one experiment, you can generate thousands of genotypes that are seen in cancer patients, and immediately test whether one or more of those genotypes are sensitive or resistant to any type of therapy that you’re interested in using,” says Francisco Sanchez-Rivera, an MIT assistant professor of biology, a member of the Koch Institute for Integrative Cancer Research, and the senior author of the study.
MIT graduate student Samuel Gould is the lead author of the paper, which appears today in Nature Biotechnology.
Editing cells
The new technique builds on research that Sanchez-Rivera began 10 years ago as an MIT graduate student. At that time, working with Tyler Jacks, the David H. Koch Professor of Biology, and then-postdoc Thales Papagiannakopoulos, Sanchez-Rivera developed a way to use CRISPR genome-editing to introduce into mice genetic mutations linked to lung cancer.
In that study, the researchers showed that they could delete genes that are often lost in lung tumor cells, and the resulting tumors were similar to naturally arising tumors with those mutations. However, this technique did not allow for the creation of point mutations (substitutions of one nucleotide for another) or insertions.
“While some cancer patients have deletions in certain genes, the vast majority of mutations that cancer patients have in their tumors also include point mutations or small insertions,” Sanchez-Rivera says.
Since then, David Liu, a professor in the Harvard University Department of Chemistry and Chemical Biology and a core institute member of the Broad Institute, has developed new CRISPR-based genome editing technologies that can generate additional types of mutations more easily. With base editing, developed in 2016, researchers can engineer point mutations, but not all possible point mutations. In 2019, Liu, who is also an author of the Nature Biotechnology study, developed a technique called prime editing, which enables any kind of point mutation to be introduced, as well as insertions and deletions.
“Prime editing in theory solves one of the major challenges with earlier forms of CRISPR-based editing, which is that it allows you to engineer virtually any type of mutation,” Sanchez-Rivera says.
When they began working on this project, Sanchez-Rivera and Gould calculated that if performed successfully, prime editing could be used to generate more than 99 percent of all small mutations seen in cancer patients.
However, to achieve that, they needed to find a way to optimize the editing efficiency of the CRISPR-based system. The prime editing guide RNAs (pegRNAs) used to direct CRISPR enzymes to cut the genome in certain spots have varying levels of efficiency, which leads to “noise” in the data from pegRNAs that simply aren’t generating the correct target mutation. The MIT team devised a way to reduce that noise by using synthetic target sites to help them calculate how efficiently each guide RNA that they tested was working.
“We can design multiple prime-editing guide RNAs with different design properties, and then we get an empirical measurement of how efficient each of those pegRNAs is. It tells us what percentage of the time each pegRNA is actually introducing the correct edit,” Gould says.
Analyzing mutations
The researchers demonstrated their technique using p53, a gene that is mutated in more than half of all cancer patients. From a dataset that includes sequencing information from more than 40,000 patients, the researchers identified more than 1,000 different mutations that can occur in p53.
“We wanted to focus on p53 because it’s the most commonly mutated gene in human cancers, but only the most frequent variants in p53 have really been deeply studied. There are many variants in p53 that remain understudied,” Gould says.
Using their new method, the researchers introduced p53 mutations in human lung adenocarcinoma cells, then measured the survival rates of these cells, allowing them to determine each mutation’s effect on cell fitness.
Among their findings, they showed that some p53 mutations promoted cell growth more than had been previously thought. These mutations, which prevent the p53 protein from forming a tetramer — an assembly of four p53 proteins — had been studied before, using a technique that involves inserting artificial copies of a mutated p53 gene into a cell.
Those studies found that these mutations did not confer any survival advantage to cancer cells. However, when the MIT team introduced those same mutations using the new prime editing technique, they found that the mutation prevented the tetramer from forming, allowing the cells to survive. Based on the studies done using overexpression of artificial p53 DNA, those mutations would have been classified as benign, while the new work shows that under more natural circumstances, they are not.
“This is a case where you could only observe these variant-induced phenotypes if you’re engineering the variants in their natural context and not with these more artificial systems,” Gould says. “This is just one example, but it speaks to a broader principle that we’re going to be able to access novel biology using these new genome-editing technologies.”
Because it is difficult to reactivate tumor suppressor genes, there are few drugs that target p53, but the researchers now plan to investigate mutations found in other cancer-linked genes, in hopes of discovering potential cancer therapies that could target those mutations. They also hope that the technique could one day enable personalized approaches to treating tumors.
“With the advent of sequencing technologies in the clinic, we’ll be able to use this genetic information to tailor therapies for patients suffering from tumors that have a defined genetic makeup,” Sanchez-Rivera says. “This approach based on prime editing has the potential to change everything.”
The research was funded, in part, by the National Institute of General Medical Sciences, an MIT School of Science Fellowship in Cancer Research, a Howard Hughes Medical Institute Hanna Gray Fellowship, the V Foundation for Cancer Research, a National Cancer Institute Cancer Center Support Grant, the Ludwig Center at MIT, a Koch Institute Frontier Award, the MIT Research Support Committee, and the Koch Institute Support (core) Grant from the National Cancer Institute.
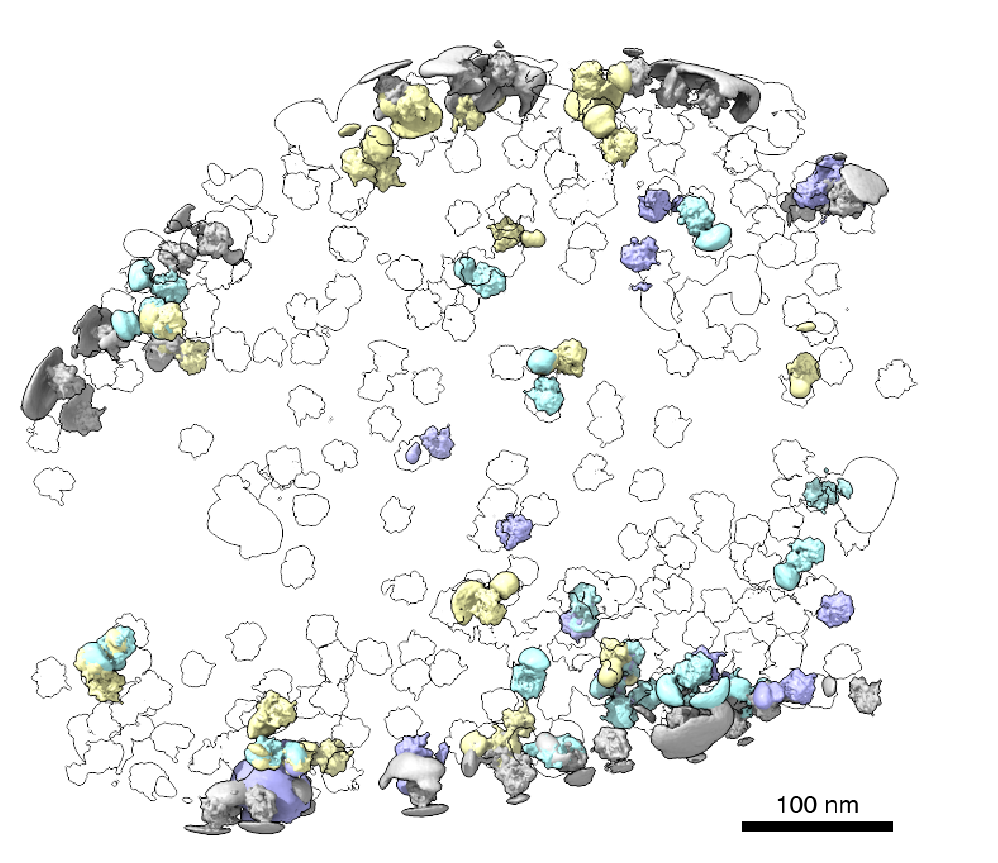
Cells rely on complex molecular machines composed of protein assemblies to perform essential functions such as energy production, gene expression, and protein synthesis. To better understand how these machines work, scientists capture snapshots of them by isolating proteins from cells and using various methods to determine their structures. However, isolating proteins from cells also removes them from the context of their native environment, including protein interaction partners and cellular location.
Recently, cryogenic electron tomography (cryo-ET) has emerged as a way to observe proteins in their native environment by imaging frozen cells at different angles to obtain three-dimensional structural information. This approach is exciting because it allows researchers to directly observe how and where proteins associate with each other, revealing the cellular neighborhood of those interactions within the cell.
With the technology available to image proteins in their native environment, graduate student Barrett Powell wondered if he could take it one step further: what if molecular machines could be observed in action? In a paper published today in Nature Methods, Powell describes the method he developed, called tomoDRGN, for modeling structural differences of proteins in cryo-ET data that arise from protein motions or proteins binding to different interaction partners. These variations are known as structural heterogeneity.
Although Powell had joined the Davis Lab as an experimental scientist, he recognized the potential impact of computational approaches in understanding structural heterogeneity within a cell. Previously, the Davis Lab developed a related methodology named cryoDRGN to understand structural heterogeneity in purified samples. As Powell and Associate Professor of Biology Joey Davis saw cryo-ET rising in prominence in the field, Powell took on the challenge of reimagining this framework to work in cells.
When solving structures with purified samples, each particle is imaged only once. By contrast, cryo-ET data is collected by imaging each particle more than 40 times from different angles. That meant tomoDRGN needed to be able to merge the information from more than 40 images, which was where the project hit a roadblock: the amount of data led to an information overload.
To address the information overload, Powell successfully rebuilt the cryoDRGN model to prioritize only the highest-quality data. When imaging the same particle multiple times, radiation damage occurs. The images acquired earlier, therefore, tend to be of higher quality because the particles are less damaged.
“By excluding some of the lower quality data, the results were actually better than using all of the data–and the computational performance was substantially faster,” Powell says.
Just as Powell was beginning work on testing his model, he had a stroke of luck: the authors of a groundbreaking new study that visualized, for the first time, ribosomes inside cells at near-atomic resolution, shared their raw data on the Electric Microscopy Public Image Archive (EMPIAR). This dataset was an exemplary test case for Powell, through which he demonstrated that tomoDRGN could uncover structural heterogeneity within cryo-ET data.
According to Powell, one exciting result is what tomoDRGN found surrounding a subset of ribosomes in the EMPIAR dataset. Some of the ribosomal particles were associated with a bacterial cell membrane and engaged in a process called cotranslational translocation. This occurs when a protein is being simultaneously synthesized and transported across a membrane. Researchers can use this result to make new hypotheses about how the ribosome functions with other protein machinery integral to transporting proteins outside of the cell, now guided by a structure of the complex in its native environment.
After seeing that tomoDRGN could resolve structural heterogeneity from a structurally diverse dataset, Powell was curious: how small of a population could tomoDRGN identify? For that test, he chose a protein named apoferritin which is a commonly used benchmark for cryo-ET and is often treated as structurally homogeneous. Ferritin is a protein used for iron storage and is referred to as apoferritin when it lacks iron.
Surprisingly, in addition to the expected particles, tomoDRGN revealed a minor population of ferritin particles–with iron bound–making up just 2% of the dataset that was not previously reported. This result further demonstrated tomoDRGN’s ability to identify structural states that occur so infrequently that they would be averaged out with traditional analysis tools.
Powell and other members of the Davis Lab are excited to see how tomoDRGN can be applied to further ribosomal studies and to other systems. Davis works on understanding how cells assemble, regulate, and degrade molecular machines, so the next steps include exploring ribosome biogenesis within cells in greater detail using this new tool.
“What are the possible states that we may be losing during purification?” Davis says. “Perhaps more excitingly, we can look at how they localize within the cell and what partners and protein complexes they may be interacting with.”