Computational neuroscientist Sven Dorkenwald and cell biologist Whitney Henry, along with two MIT alumni, are recognized for their exceptional early-career research contributions.
Julie Pryor | Bendta Schroeder | McGovern Institute for Brain Research | Koch Institute
May 20, 2026
MIT scientists Sven Dorkenwald and Whitney Henry have been named 2026 Searle Scholars, an award given annually to 15 exceptional early-career researchers in the fields of biomedical sciences and chemistry. Dorkenwald is an assistant professor of brain and cognitive sciences and an investigator at the McGovern Institute for Brain Research. Henry is the Robert A. Swanson (1969) Career Development Professor of Life Sciences and an intramural faculty member at the Koch Institute for Integrative Cancer Research.
In addition, MIT alumni Irene Kaplow ’10 and Jared Mayers PhD ’15 were also honored.
Chosen by a scientific advisory board, Searle Scholars are considered among the most creative young researchers pursuing high-risk/high-reward research. The Searle Scholars Program is funded through the Searle Funds at The Chicago Community Trust and administered by Kinship Foundation. Each scholar will each receive $450,000 in flexible funding to support their work over the next three years.
Sven Dorkenwald
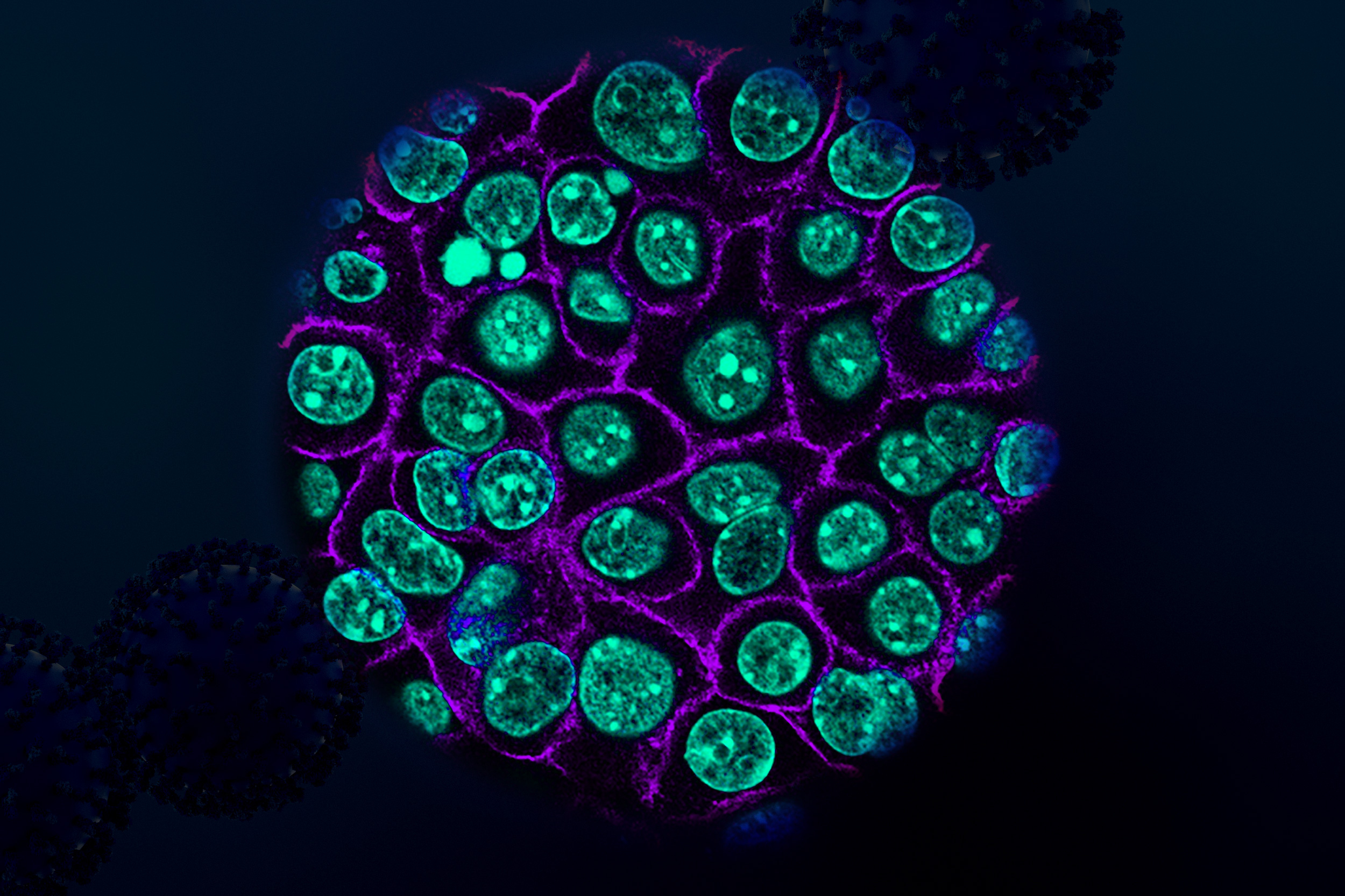
Sven Dorkenwald is a computational neuroscientist investigating the organizational principles of neuronal circuits. The synaptic connectivity of neurons, their connectome, is fundamental to how networks of neurons function. Dorkenwald develops computational and collaborative tools to map, analyze, and interpret synapse-resolution connectomes. His work has led to large connectomic reconstructions of the fruit fly brain and parts of mammalian brains. He uses these connectomes to investigate the architecture of neuronal circuits and how their structure supports complex computations.
“As I establish my new lab, the Searle Scholars Award will help us launch ambitious projects and set our long-term scientific direction,” says Dorkenwald. “I am deeply grateful for the support from the Kinship Foundation and look forward to interacting with this amazing cohort of Searle Scholars.”
Dorkenwald joined the faculty of MIT in 2026 as an assistant professor in the Department of Brain and Cognitive Sciences and an investigator at the McGovern Institute. He earned a BS in physics and an MS in computer engineering from the University of Heidelberg, followed by a PhD in computer science and neuroscience at Princeton University in 2023 under the mentorship of Sebastian Seung and Mala Murthy. Dorkenwald completed his postdoctoral training as a Shanahan Research Fellow at the Allen Institute and the University of Washington, while serving as a visiting faculty researcher at Google Research.
Whitney Henry
Whitney Henry investigates the potential of ferroptosis, an iron-dependent form of cell death, for developing novel therapies that target subpopulations of cancer cells that are highly metastatic, therapy-resistant, and therefore critical instigators of tumor relapse. Her research is focused on uncovering the molecular factors influencing ferroptosis susceptibility, investigating its effects on the tumor microenvironment, and developing innovative methods to manipulate ferroptosis resistance in living organisms, drawing from functional genomics, metabolomics, bioengineering, and a range of in vitro and in vivo models.
“I am incredibly grateful to the Kinship Foundation for supporting our research and giving us the freedom to ask bold, curiosity-driven scientific questions,” says Henry. “This support allows us to pursue ambitious ideas, take creative risks, and embark on new research directions.”
Henry joined the MIT faculty in 2024 as an assistant professor in the Department of Biology and a member of the Koch Institute, and is currently an HHMI Freeman Hrabowski Scholar. She received her bachelor’s degree in biology with a minor in chemistry from Grambling State University and her PhD from Harvard University. Following her doctoral studies, she worked in the lab of Robert Weinberg at the Whitehead Institute for Biomedical Research and was supported by fellowships from the Jane Coffin Childs Memorial Fund for Medical Research and the Ludwig Center at MIT.
Alumni also honored
Irene Kaplow ’10, a graduate of the MIT Department of Mathematics, is an assistant professor in the Department of Biology and the Ray and Stephanie Lane Computational Biology Department at Carnegie Mellon University. Her selection as a Searle Scholar is for “deciphering transcriptional regulatory mechanisms underlying mammalian dietary phenotype evolution and their relationships to transcriptional regulatory responses to changes in diet.”
Jared Mayers PhD ’15, who earned his doctorate from the MIT Department of Biology, is an assistant professor at the Fred Hutchinson Cancer Center at the University of Washington. His selection as a Searle Scholar is for “a reverse-translational framework to decipher metabolic vulnerabilities of bacterial pathogens.”