Biology PhD student Giselle Valdes (Reddien Lab) studies stem cell regeneration while encouraging aspiring students and researchers.
Stefanie Koperniak | Division of Graduate and Undergraduate Education
June 11, 2026
As an undergraduate at Florida International University, Giselle Valdes tackled rigorous studies in the school’s Honors College while simultaneously caring for family members with medical needs.
“I think that the choice to pursue any field in the space of biology and medical research was entirely shaped by having to be there for my family,” says Valdes.
As a McNair Scholar and biomedical engineering major who also did extensive research in biochemistry, she leaned more toward undergraduate courses in mechanical and electrical engineering that were geared primarily toward equipping students to build medical devices. She began to shift her research interests more firmly into biology, however, the summer before her senior year in 2018. She spent 10 weeks on the MIT campus as a participant in the Bernard S. and Sophie G. Gould MIT Summer Research Program in Biology (BSG-MSRP-Bio), working in the lab of Associate Professor Eliezer Calo PhD ’11, also a former BSG-MSRP-Bio participant. The Calo Lab focuses on ribosomes, small cellular particles that translate RNA into proteins, and looks at how mutations in ribosome development can lead to disorders.
After working in Calo’s lab, she could see herself as a biology graduate student at MIT. In January 2019, she attended the MIT biology department’s Quantitative Methods Workshop, a weeklong, intensive workshop designed to introduce non-MIT undergraduates to tools and programming languages used to analyze experimental data in biology and neuroscience. While there, she was elated to receive an email from the department inviting her to interview for the PhD program. She was accepted and began her doctoral studies in the fall of that year.
“When I think about my experiences at MIT, both as an undergraduate in MIT programs and as a PhD candidate in biology, I think about all the great mentors who have helped me along the way,” says Valdes. “I’ve also really valued the richly collaborative community, and being able to take a lot of risks in how I address the questions I have the opportunity to pursue.”
Researching stem cell regeneration
Since she came from a biomedical engineering background, Valdes spent the first year of the biology doctoral program taking foundational biology courses and working in different labs to decide which type of research she wanted to do. She gravitated toward cell and developmental biology and joined the lab of Professor Peter Reddien, associate director of the Whitehead Institute for Biomedical Research. Valdes was awarded an MIT Fund for the Future of Science Fellowship to support her research.
“Giselle is doing terrific work on a fundamental problem related to adult stem cells and regeneration — how do progenitor cells choose what cell types to make? Fate choice in progenitors is typically studied in embryogenesis, and how it occurs in the context of adult regeneration is poorly understood and very important to address,” says Reddien.
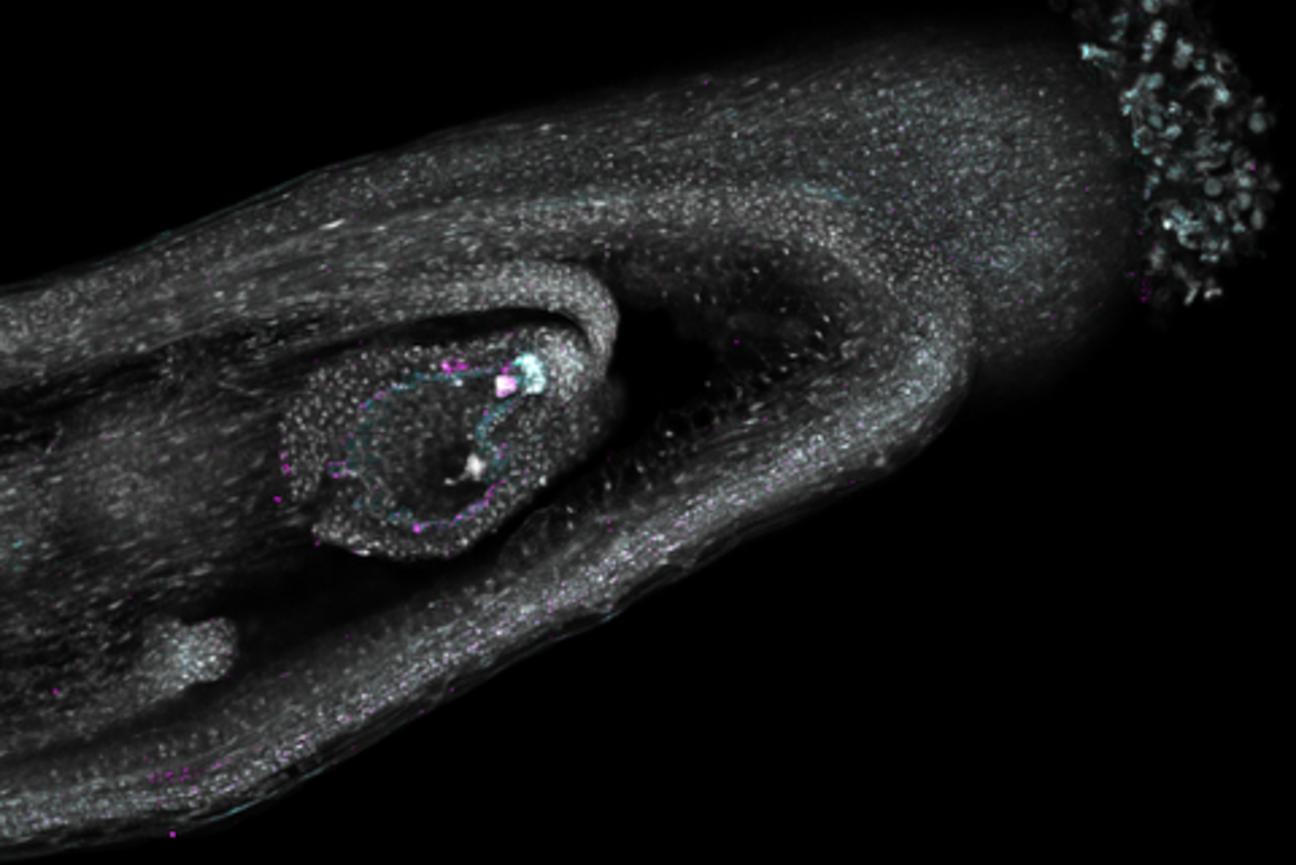
Valdes has worked extensively with stem cells in highly regenerative flatworms, called planaria. analyzing the process of “cell fate choice,” or how cells determine which specific cell types and functions to develop. To date, Valdes, Reddien, and other researchers have studied “neighborhoods” of neoblasts (adult stem cells) and their fate choices, finding that different neighboring stem cells often chose different fate options — suggesting that cell fate choices are largely made by processes autonomous to individual cells.
Her current research aims to better understand the driving mechanism for cell fate choice, both within planaria and an additional model system: the evolutionarily distant acoel Hofstenia miamia.
“A lot of the things I’m doing in my current project have involved developing techniques that didn’t previously exist in our model organism,” says Valdes.
Working on model systems with limitations in the toolkit traditionally available to more well-established systems, such as transgenics, has allowed her to be creative in the techniques she applies to determine how stem cells choose what to become. It has also opened doors to collaborations, such as one with Ye Zhang of the Manalis Lab in the biological engineering department (now an assistant professor of biomedical engineering at Virginia Tech), that have allowed Valdes and team to sort neoblasts in novel ways based on their morphology, and better relate that to their dynamic state.
In summer 2024, Valdes mentored a BSG-MSRP-Bio student who now works with her on a current research project.
“She’s been with me as a technical assistant in the lab now for over a year, and we’ve been able to work on one of my projects together,” says Valdes. “It’s been exciting to come full circle in this way.”
Teaching and mentoring, near and far
In addition to her research, Valdes devotes a lot of her time to teaching and mentoring, both for MIT biology students and younger students discovering an interest in STEM.
“It’s been so rewarding to have a lot of opportunities to do for others what has been done for me,” she says.
Valdes has worked with secondary students both locally and abroad. She participates in the biology department’s developmental biology lab for high-school students and teaches in an annual biology lecture series for high schoolers. She has worked with the Enroot program from Cambridge Community Services, acting as a direct mentor to local high-school and community college students. At the Whitehead Institute’s Expedition: Bio program, for middle- and high-school students, she runs a planarian workshop. And she gives lab tours through the Whitehead Discovery Lab initiative, engaging in discussion with local high-school students.
Valdes has also assisted with a hackathon for Sprouting, a social impact venture providing STEM education opportunities to under-resourced communities in Puerto Rico. Sprouting was launched by Taylor Baum, a doctoral student in MIT’s Department of Electrical Engineering and Computer Science. Valdes taught coding essentials to Spanish-speaking middle- and high-school students in Puerto Rico.
“That was really emotional,” says Valdes. “The parents were so grateful, and there were kids who were clearly brilliant and gifted. They were able to really take off with the tools that we gave them.”
In her department, Valdes has been a teaching assistant for classes 7.003 (Applied Molecular Biology Laboratory) and 7.03 (Genetics). She is also a teaching assistant for the Quantitative Methods Workshop and teaches a Python module to students in the program.
“Giselle may be quite small in physical stature, yet she dominates the room when she speaks, and commands the full attention of an audience of 80 students when giving a lecture,” says Mandana Sassanfar, a biology senior lecturer and director of outreach who runs the Quantitative Methods Workshop. “She is highly respected both for her knowledge and the way she interacts with people. She is extremely approachable, very generous with her time, and always very supportive and encouraging. She is a wonderful mentor, teacher, and scientist.”
Valdes says she is always happy to help mentor undergrads and graduate students. She is co-founder and coordinator of the MIT Biology Application Assistance Program (BAAP), which aims to demystify the graduate school application process and offer interested applicants the tools and direct mentorship necessary for putting together a successful application. She also helped to coordinate, and has been an active participant in, the MIT BioPals Program, a student-organized peer mentorship initiative within the department that connects incoming first-year graduate students with senior graduate students. During the Covid-19 pandemic, this program provided critical support and social connection for new students navigating remote learning and social distancing.
After she completes her doctoral program, she envisions pursuing a postdoc and, ultimately, a faculty role, citing her passion for both academic research and teaching.
“My goal is to stay in academia in some way,” says Valdes. “I love mentorship and curiosity-driven science.”