They identified proteins that influence splicing of about half of all human introns, allowing for more complex types of gene regulation.
Anne Trafton | MIT News
February 20, 2025
RNA splicing is a cellular process that is critical for gene expression. After genes are copied from DNA into messenger RNA, portions of the RNA that don’t code for proteins, called introns, are cut out and the coding portions are spliced back together.
This process is controlled by a large protein-RNA complex called the spliceosome. MIT biologists have now discovered a new layer of regulation that helps to determine which sites on the messenger RNA molecule the spliceosome will target.
The research team discovered that this type of regulation, which appears to influence the expression of about half of all human genes, is found throughout the animal kingdom, as well as in plants. The findings suggest that the control of RNA splicing, a process that is fundamental to gene expression, is more complex than previously known.
“Splicing in more complex organisms, like humans, is more complicated than it is in some model organisms like yeast, even though it’s a very conserved molecular process. There are bells and whistles on the human spliceosome that allow it to process specific introns more efficiently. One of the advantages of a system like this may be that it allows more complex types of gene regulation,” says Connor Kenny, an MIT graduate student and the lead author of the study.
Christopher Burge, the Uncas and Helen Whitaker Professor of Biology at MIT, is the senior author of the study, which appears today in Nature Communications.
Building proteins
RNA splicing, a process discovered in the late 1970s, allows cells to precisely control the content of the mRNA transcripts that carry the instructions for building proteins.
Each mRNA transcript contains coding regions, known as exons, and noncoding regions, known as introns. They also include sites that act as signals for where splicing should occur, allowing the cell to assemble the correct sequence for a desired protein. This process enables a single gene to produce multiple proteins; over evolutionary timescales, splicing can also change the size and content of genes and proteins, when different exons become included or excluded.
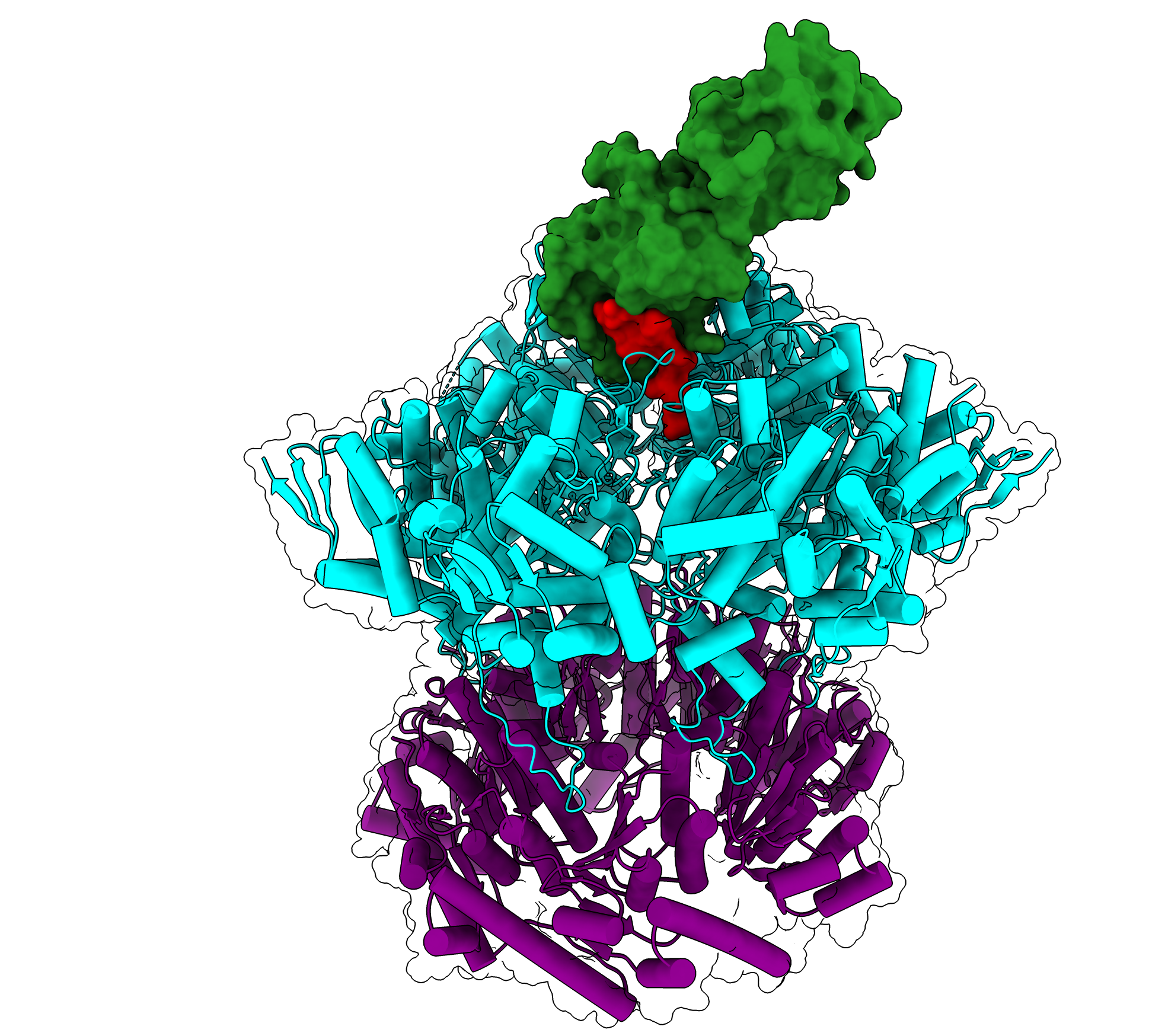
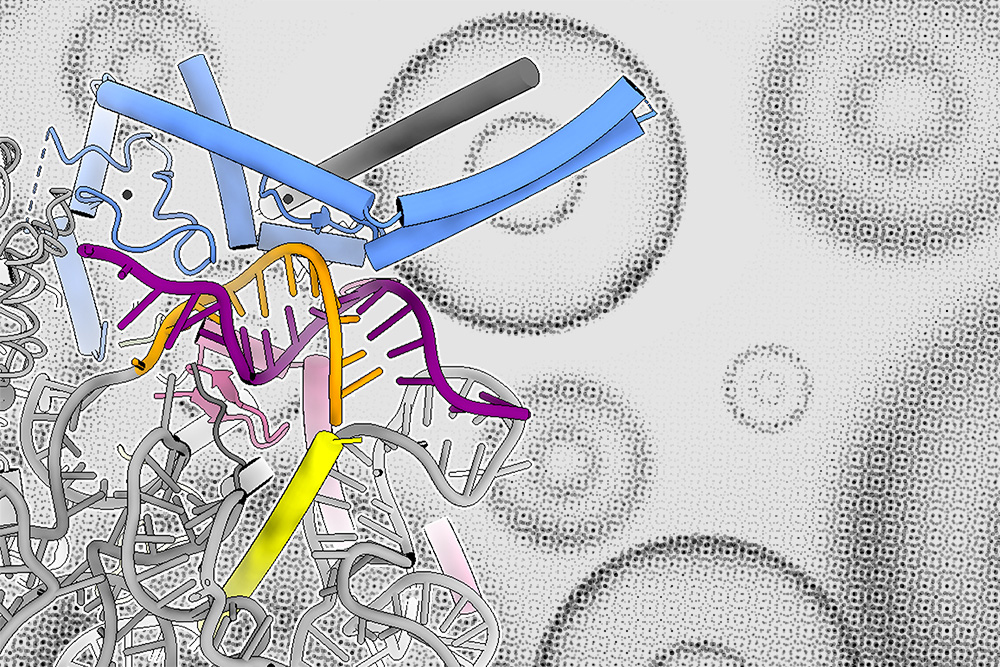
The spliceosome, which forms on introns, is composed of proteins and noncoding RNAs called small nuclear RNAs (snRNAs). In the first step of spliceosome assembly, an snRNA molecule known as U1 snRNA binds to the 5’ splice site at the beginning of the intron. Until now, it had been thought that the binding strength between the 5’ splice site and the U1 snRNA was the most important determinant of whether an intron would be spliced out of the mRNA transcript.
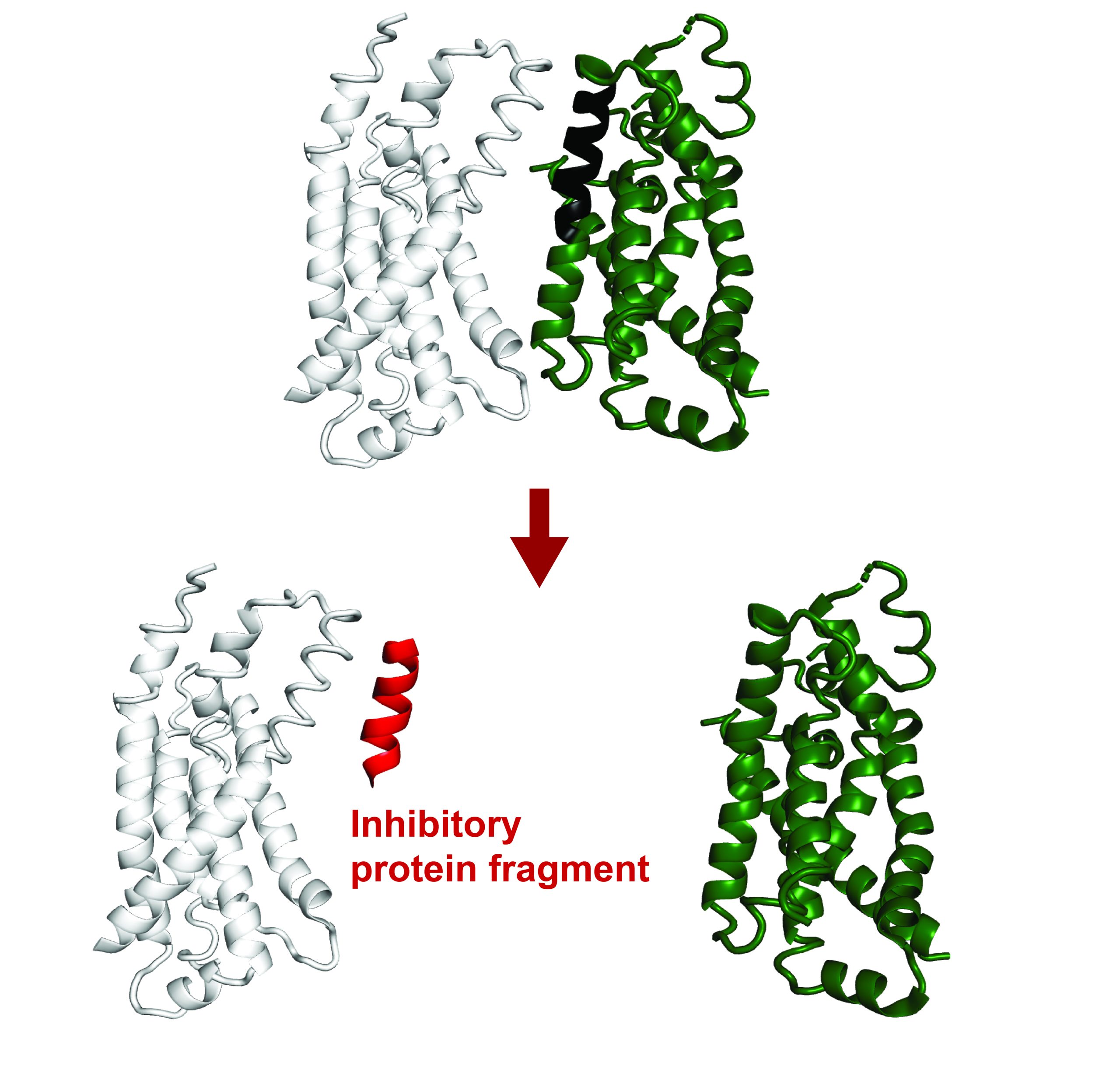
In the new study, the MIT team discovered that a family of proteins called LUC7 also helps to determine whether splicing will occur, but only for a subset of introns — in human cells, up to 50 percent.
Before this study, it was known that LUC7 proteins associate with U1 snRNA, but the exact function wasn’t clear. There are three different LUC7 proteins in human cells, and Kenny’s experiments revealed that two of these proteins interact specifically with one type of 5’ splice site, which the researchers called “right-handed.” A third human LUC7 protein interacts with a different type, which the researchers call “left-handed.”
The researchers found that about half of human introns contain a right- or left-handed site, while the other half do not appear to be controlled by interaction with LUC7 proteins. This type of control appears to add another layer of regulation that helps remove specific introns more efficiently, the researchers say.
“The paper shows that these two different 5’ splice site subclasses exist and can be regulated independently of one another,” Kenny says. “Some of these core splicing processes are actually more complex than we previously appreciated, which warrants more careful examination of what we believe to be true about these highly conserved molecular processes.”
“Complex splicing machinery”
Previous work has shown that mutation or deletion of one of the LUC7 proteins that bind to right-handed splice sites is linked to blood cancers, including about 10 percent of acute myeloid leukemias (AMLs). In this study, the researchers found that AMLs that lost a copy of the LUC7L2 gene have inefficient splicing of right-handed splice sites. These cancers also developed the same type of altered metabolism seen in earlier work.
“Understanding how the loss of this LUC7 protein in some AMLs alters splicing could help in the design of therapies that exploit these splicing differences to treat AML,” Burge says. “There are also small molecule drugs for other diseases such as spinal muscular atrophy that stabilize the interaction between U1 snRNA and specific 5’ splice sites. So the knowledge that particular LUC7 proteins influence these interactions at specific splice sites could aid in improving the specificity of this class of small molecules.”
Working with a lab led by Sascha Laubinger, a professor at Martin Luther University Halle-Wittenberg, the researchers found that introns in plants also have right- and left-handed 5’ splice sites that are regulated by Luc7 proteins.
The researchers’ analysis suggests that this type of splicing arose in a common ancestor of plants, animals, and fungi, but it was lost from fungi soon after they diverged from plants and animals.
“A lot what we know about how splicing works and what are the core components actually comes from relatively old yeast genetics work,” Kenny says. “What we see is that humans and plants tend to have more complex splicing machinery, with additional components that can regulate different introns independently.”
The researchers now plan to further analyze the structures formed by the interactions of Luc7 proteins with mRNA and the rest of the spliceosome, which could help them figure out in more detail how different forms of Luc7 bind to different 5’ splice sites.
The research was funded by the U.S. National Institutes of Health and the German Research Foundation.