Research Area: Microbiology

Education
- Graduate: PhD, 2011, MIT; MD, 2013, Harvard Medical School
- Undergraduate: BA, 2006, Biology, University of Chicago
Research Summary
Diverse commensal microbes colonize every surface of our bodies. We study the constant communication between these microbes and our immune system. We focus on our largest organ: the skin. By employing microbial genetics, immunologic approaches, and mouse models, we can dissect (1) the molecular signals used by microbes to educate our immune system and (2) how different microbial communities alter immune responses. Ultimately, we aim to harness these microbe-host interactions to engineer novel vaccines and therapeutics for human disease.
Awards
- Howard Hughes Medical Institute Hanna H. Gray Fellow, 2018-2026
- A.P. Giannini Postdoctoral Research Fellowship, 2018
- Dermatology Foundation Research Fellowship, 2017
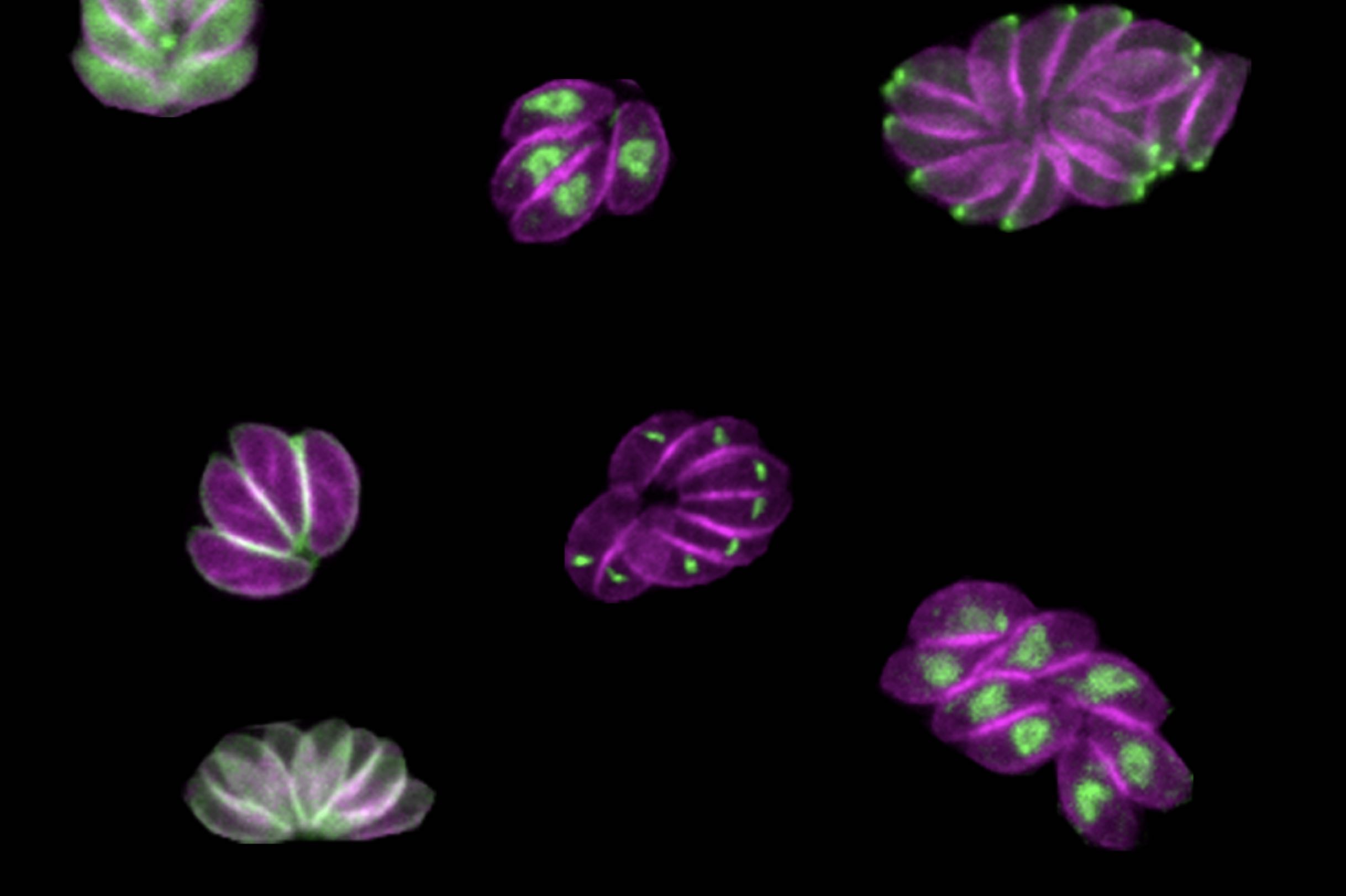
Eva Frederick | Whitehead Institute
April 28, 2022
Whitehead Institute Member Sebastian Lourido and his lab members study the parasite Toxoplasma gondii. The parasite causes the disease toxoplasmosis, which can be dangerous for pregnant or immunocompromised patients.
As the parasite evolved over millennia, its phylum (the Apicomplexan parasites) split off from other branches of life, which poses a challenge to researchers hoping to understand its genetics. “Toxoplasma is very highly diverged from the organisms that we typically study, like mice, yeast and [nematodes],” said Lourido lab researcher and Massachusetts Institute of Technology (MIT) graduate student Tyler Smith. “Our lab focuses a lot on developing toolkits to probe and study the genomes of these parasites.”
Now, in a paper published in the journal Nature Microbiology on April 28, Smith and colleagues describe a new method for determining the role of genes within the genome of the parasite. The method can be conducted by a single investigator, and goes a step beyond simply assessing whether or not a given gene is essential for survival. By inserting specific sequences — such as those encoding fluorescent markers or sequences that can turn a gene on and off — throughout the Toxoplasma genome, the method allows the researchers to visualize where an individual gene’s product resides within the parasites and identify when in the life cycle important genes became essential, providing more detailed information than a traditional CRISPR screen.
Although the method could theoretically be used with any gene family, Smith and Lourido decided to first focus on a family of proteins called kinases, the genetic code for which comprises around 150 of Toxoplasma’s 8,000 total genes.
“Kinases are interesting from a basic biology perspective because they are signaling hubs of basic biological processes,” said Smith, who is first author of the study. “From a more translational perspective, kinases are really common drug targets. We have a lot of inhibitors that work with kinases. For some cancers that are linked to specific kinases, the inhibitors can be chemotherapies.”
Using the method, researchers discovered a gene encoding a previously unstudied kinase which they named SPARK. They were able to show that the SPARK kinase is involved in the process of the parasites entering and leaving host cells, and future research on inhibitors of SPARK could lead to new treatments for toxoplasmosis. “Identifying these kinases that are really vital for these critical decision points in a parasite’s life cycle could be really fruitful for developing new therapeutics,” said Lourido, who is also an associate professor of biology at MIT.
New dimensions of screening
Many CRISPR screens use gene editing technology to knock out genes throughout the genomes of a sample of cells, creating a population where every gene in the genome is mutated in at least one of the cells. Then, by looking at which mutations have detrimental effects on the cells, researchers can extrapolate which genes are essential for survival.
But the workings of a whole organism are infinitely more complicated than just survival or death, and researchers are often faced with a challenge when it comes to figuring out exactly what different gene products are doing in the cells. That’s why Smith and Lourido decided to design a method of screening for Toxoplasma genes that could provide more information about what the products of those genes do. “CRISPR screens can tell you which genes are important, but it doesn’t give you much information about why they’re important,” Smith said. “We were seeking to make a kind of platform that could look at other dimensions.”
Smith and Lourido used CRISPR technology to introduce small amounts of new DNA into the parasites’ genes that code for kinases. The new DNA included sequences encoding a fluorescent marker protein and sequences that could be used to manipulate gene expression levels.
After creating a population of parasites modified this way, the researchers then used imaging to determine where the fluorescently tagged proteins had ended up in the cells, and to observe what happened in the cells when the proteins were turned off. “Being able to see different cell division phenotypes — for instance parasites that either failed to replicate at all, or tried to replicate but would have some abnormalities — that gets us closer and allows us to generate hypotheses as to actually why these kinases are important, not just whether or not they are important,” Smith said.
The depletion of some proteins caused the parasites to die instantly, while others affected the parasites at a later point in their life cycles, so they would drop out of the population more slowly. “Cells with mutations in these kinases replicate fine, but a problem might arise when they need to leave their host cell and enter a new host cell later on down the line,” Smith said.
A “SPARK” of inspiration
After the screen, the researchers followed up on one of these kinases in particular, which they called SPARK (short for Store Potentiating/Activating Regulatory Kinase). Mutants depleted of SPARK died, but not until a later phase of the life cycle. Smith and Lourido conducted further experiments to understand SPARK’s role, and found evidence that the protein was involved in the release of calcium in the cell that is required for a parasite to enter or leave a host cell.
“The thing I found very interesting about SPARK is that it’s a kinase that’s very different from the analogous kinase in other model organisms, but is conserved throughout all of the apicomplexan phylum,” Smith said. “That’s the phylum that includes Toxoplasma and a bunch of other single-celled parasites like Plasmodium, which is the malaria parasite.”
Because SPARK is far different from its human analog and essential to the parasite’s life cycle, a SPARK-specific kinase inhibitor could be used to treat toxoplasmosis by killing the parasite without affecting the patient. “The hope would be that you can target SPARK and inhibit it without hitting mammalian kinases,” Smith said. “It’s easy enough to design something that kills a cell, but the trick is only killing parasites and not your own cells.”
In the future, the researchers hope to turn their new screening method to other families of genes, such as transcription factors, to understand their function in the parasites. “Our results have been quite encouraging in that we think this method will be scalable, and we can target larger gene sets in the future,” Smith said. “I think the ultimate end goal would be to do the whole genome.”
“There’s this whole universe of parasite proteins that we know so little about, where this type of analysis will be incredibly insightful.” Lourido said. “We’re really very excited about scaling it up further.

Graduate student En Ze Linda Zhong-Johnson is creating new methods to measure and enhance enzyme activity — which she hopes will help restore a plastic-choked world.
Grace van Deelen
April 21, 2022
After graduating with her undergraduate degree in molecular genetics from the University of Toronto in 2016, En Ze Linda Zhong-Johnson celebrated with a trip to Alaska. There, she saw a pristine landscape unlike the plastic-littered shores of the Toronto waterfront. “What I saw up there was so different from what I saw in the city,” Zhong-Johnson says. “I realized there shouldn’t be all this waste floating everywhere, in our water, in our environment. It’s not natural.”
As a trained biologist, Zhong-Johnson began to think about the problem of plastic pollution from a biological perspective. One solution, she thought, could be biological recycling: a process by which living organisms break down materials, using digestion or other metabolic processes to turn these materials into smaller pieces or new compounds. Composting, for example, is a type of biological recycling — microbes in the soil break down discarded food, speeding up the decomposition process. Zhong-Johnson wondered if there were any organisms on Earth that could use the carbon in polyethylene terephthalate (PET), a common plastic used in water bottle and food packaging, as an energy source.
Earlier that same year, Japanese scientists discovered that a bacterium, Ideonella sakaiensis, could do just that by producing enzymes that could break down PET. The two main PET-degrading enzymes, referred to as IsPETase and IsMHETase, are able to turn PET into two chemical compounds, terephthalic acid and ethylene glycol, which I. sakaiensis can use for food.
The discovery of these enzymes opened up many new questions and possible applications that scientists have continued to work on since. However, because there was — and still is — much to learn about PET-degrading enzymes, they are still not widely used to recycle consumer products. Zhong-Johnson figured that, in graduate school, she could build on the existing IsPETase research and help to accelerate their use at recycling facilities. Specifically, she wanted to engineer the enzyme to work faster at lower temperatures, and study how, fundamentally, the enzymes worked on the surface of PET plastic to degrade it.
“I hopped on the excitement train, along with the rest of the world,” she says.
A better enzyme
After receiving her acceptance to MIT to complete her PhD, Zhong-Johnson approached various professors, pitching her idea to speed up IsPETase activity. Christopher Voigt, the Daniel I. C. Wang Professor of Biological Engineering, and Anthony Sinskey, professor of biology, were interested, and formed a co-advisorship to support Zhong-Johnson’s project. Sinskey, in particular, was impressed by her idea to help solve the world’s plastic problem with PET-degrading enzymes.

“Plastic pollution is a big problem,” he says, “and that’s the kind of problem my lab likes to tackle.” Plus, he says, he feels “committed to helping graduate students who want to apply their science and technology learnings to the environment.”
While the idea of a plastic-degrading enzyme seems like a panacea, the enzyme’s practical applications have been limited by its biology. The wild-type IsPETase is a mesophilic enzyme, meaning the structure of the enzyme is only stable around ambient temperatures, and the enzyme loses its activity above that threshold. This restriction on temperature limits the number and types of facilities that can use IsPETase, as well as the rate of the enzyme reaction, and drives up the cost of their use.
However, Zhong-Johnson thinks that, with combined approaches of biological and chemical engineering, it’s possible to scale up the use of the enzymes by increasing their stability and activity. For example, an enzyme that’s highly active at lower temperatures could work in unheated facilities, or even be sprinkled directly into landfills or oceans to degrade plastic waste — a process called bioremediation. Increasing the activity of the enzyme at ambient temperatures could also expand the possible applications.
“Most of the environments where plastic is present are not above 50 degrees Celsius,” said Zhong-Johnson. “If we can increase enzyme activity at lower temperatures, that’s really interesting for bioremediation purposes.”
Now a fifth-year graduate student, Zhong-Johnson has honed her project, and is focusing on increasing the activity of IsPETase. To do so, she’s using directed evolution — creating random mutations in the IsPETase gene, and selecting for IsPETase variants that digest PET faster. When they do, she combines the beneficial mutations and uses that as template for the next round of library generation, to improve the enzyme even further. The evolution is “directed” because Zhong-Johnson herself, rather than nature, is picking out which gene sequences of enzyme proceed through to the next round of random mutagenesis, and which don’t. Her ultimate goal is to create a more efficient and hardier enzyme that will, hopefully, work faster at ambient temperatures.
A better protocol
Just as Zhong-Johnson was beginning her project, she ran into an obstacle: There wasn’t a standard way to measure whether her experiments were successful. In particular, no immediately applicable method existed to measure enzyme kinetics for IsPETases on solid substrates like plastic bottles and other plasticware. That was a problem for Zhong-Johnson because understanding enzyme activity was a crucial part of how she selected her enzymes in the directed evolution process.
Usually, enzyme activity is measured via product accumulation: When enzymes metabolize a substance, they create a new substance in return, called a product. Measuring the amount of product created by an enzyme after a certain amount of time gives the researcher a snapshot of that enzyme’s activity.
There are two problems with the product accumulation method, though. First, it is usually done using liquid or soluble substrates. In other words, the material that the enzyme is targeting is dissolved, like sugar dissolved in water. Then, the enzyme is added to that liquid concoction and mixed evenly throughout. However, the substrate Zhong-Johnson wanted to use — PET — was not soluble but solid, meaning it could not be evenly distributed like a soluble substrate. Second, the product accumulation measurement methods available were only practical for measuring less than a handful of timepoints for a few enzyme or substrate concentrations. As a result, many in the field opted to measure a single time point, late in the enzyme reaction, which doesn’t provide an indication of how an enzyme’s rate of digestion actually changes over the course of time — something that can be measured through kinetic measurements.
Taking kinetic measurements would help researchers like Zhong-Johnson illustrate the full pattern of enzyme activity and answer questions like: When is the enzyme most active? Does most product accumulation happen at the beginning of the reaction or the end? How does temperature impact the rate of these reactions over time? To answer these questions, she realized she would have to develop the method herself.
Through a serendipitous discussion with a group of chemical engineering undergraduate students that Zhong-Johnson was mentoring, she came up with a solution, which she published in a 2021 paper in Scientific Reports. The undergraduates brought to her attention many factors that she had overlooked about the enzyme, and she says she would not have realized the importance of kinetic measurements if it weren’t for the fact that she was trying to design an experiment that the undergraduates could perform over the course of three hours.
The paper outlined a new way to measure enzyme activity, which Zhong-Johnson calls “the bulk absorbance method.” Instead of measuring the final product accumulation at very late time points, the bulk absorbance method involves taking multiple kinetic measurements at early time intervals during the experiment. This technique informs Zhong-Johnson’s directed evolution approach: If she can find which enzymes are most active at low temperatures, she can select the best possible enzyme for the next round of analyses. She hasn’t yet engineered an enzyme she’s completely happy with, but she’s gotten much closer to her ultimate goal.
Solving big problems together
Zhong-Johnson’s discoveries have been made possible by the collaboration between her and her two co-advisors, Voigt and Sinskey, who have supported her independence throughout her five years at MIT.

When she first started her graduate work, neither Voigt nor Sinskey had expertise in enzyme biochemistry involving solid substrates: Sinkey’s lab focuses on bacterial metabolism, while Voigt’s lab focuses on genetic engineering (though Voigt did have experience with directed evolution research). Additionally, Zhong-Johnson’s path to her project was rather unconventional. Most grad students do not come to potential advisors proposing entire dissertations, which posed a unique challenge for Zhong-Johnson.
Despite not having specific expertise in enzyme biochemistry involving solid substrates, Voigt and Sinskey have supported Zhong-Johnson in other ways: by helping her to develop critical thinking skills and connecting her to other people in her field, such as potential collaborators, who can help her project thrive in the future. Zhong-Johnson has supplemented her MIT experience by having enzyme experts as part of her dissertation committee as well.
Sinskey says that, in the future — once Zhong-Johnson has engineered the ideal enzyme — they would like to partner with industry, and work on making the enzyme into a product that waste companies might use to recycle plastic. Additionally, Sinskey says, the plastic problem and the IsPETase solution raise so many interesting questions that Zhong-Johnson’s project will probably live on in the Voigt and Sinskey labs even after she graduates. He’d like to see other graduate students working to understand the enzyme’s activity and progressing the directed evolution that Zhong-Johnson started.
Zhong-Johnson is already working on understanding the specifics of how IsPETase act on PET. “How does it eat a hole in a plastic bottle? How does it move along and make the hole bigger as it moves through the process? Does it jump around? Or does it keep degrading a single polymer chain until its completely broken down? We just don’t know the answers yet,” says Sinskey.
But Zhong-Johnson is up to the task. “My graduate students have to have three skills, in my opinion,” Sinskey says. “One, they have to be intelligent. Two, they have to be energetic, and three, they have to be of high integrity, in research and behavior.” Zhong-Johnson, he says, has all three qualities.
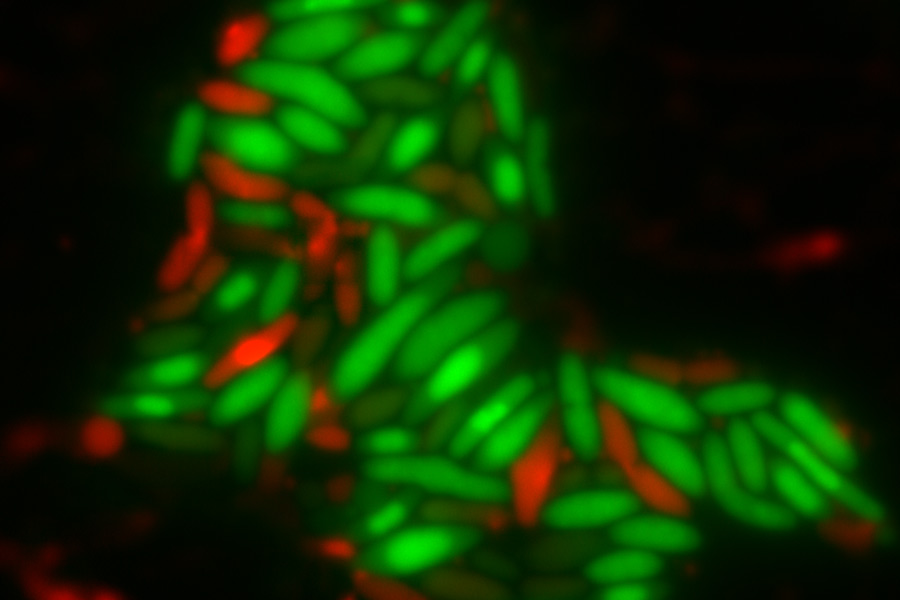
Depletion of either the DapB or Dxr proteins causes oxidative stress and cell death in bacteria, which could aid the development of more effective antibiotics.
Grace van Deelen
April 7, 2022
How do bacteria die? It’s an important question, especially since these single-celled organisms seem to be outpacing the development of new antibiotics. However, any one bacterial cell will often die from a number of separate but related pathways acting simultaneously, making understanding bacterial death difficult. Determining how to induce those pathways — and the role each pathway plays in a cell’s death — is key to creating effective antibiotics, especially after bacteria evolve to resist some drugs. But new research from Graham Walker’s lab in the MIT Department of Biology suggests that one historically under-appreciated cause of bacterial death, called oxidative stress, could help scientists develop antibiotics that kill bacteria more effectively.
Many antibiotics target the bacteria’s cell wall or replication process. However, some antibiotics can additionally cause changes in a cell’s metabolism that lead to a phenomenon called oxidative stress. Reactive oxygen-containing molecules float around freely inside the cell, sometimes bumping into other molecules, reacting with them, and stealing their unpaired electrons in a process called oxidation. For example, a guanine molecule — the DNA nucleotide commonly abbreviated to “G” — may become an oxidized guanine called 8-oxo-dG, a transformation that causes mutations in a cell’s genetic code. The harmful effects of oxidation are usually managed by the cell, but the disruption caused by these antibiotics can also become fatal to the cell.
In the case of 8-oxo-dG, the cell responds to this oxidative stress by attempting to cut the oxidized guanine out of the genome and repair it with a regular nucleotide during a process called base excision repair (BER). However, during BER, every completed step produces intermediate substances, including other forms of damaged DNA, which then must be cleared by another enzyme or protein. However, sometimes the cell is unable to complete BER because these intermediate substances build up. When there is an imbalance of intermediate substances, the cell pauses the repair, leaving breaks in the strands of DNA that cause cell death.
Because incomplete BER is just one of many contributing causes of cell death, the total contribution of incomplete BER to cell death remained unclear. As a result, scientists in the Walker lab were interested in determining other stressors, besides known antibiotics, that might cause incomplete BER of 8-oxo-dG. “If this mechanism of 8-oxo-dG getting into DNA causes bacteria to die, there’s probably some other stressor that isn’t an antibiotic that would cause cells to die by the same way,” says Walker.
In the paper, published on February 8 in mBio, the researchers determined two additional stressors that also induce cell death via incomplete BER of 8-oxo-dG. They found that the depletion of proteins DapB and Dxr also induced oxidative stress and incomplete BER of 8-oxo-dG. Scientists have known of these proteins — both of which are involved in bacterial metabolism — for some time, but had never associated them with incomplete BER.
“Incomplete base excision repair is probably one of more underappreciated ways a cell can die,” Walker says. “So we wanted to explore that pathway further.”
Charley Gruber, a postdoc in the Walker lab and lead author on the paper, identified DapB and Dxr by screening a library of 238 proteins essential for Escherichia coli growth. He determined that, in the absence of these two proteins, the cell overproduced the reactive oxygen-containing molecules that contribute to oxidative stress. As a result, the oxidized nucleotide 8-oxo-dG was incorporated into the genome, leading to cell death through incomplete BER. Researchers don’t know for sure why depletion of DapB and Dxr increases the amount of reactive oxygen-containing molecules inside the cell, but oxidative stress is a common reaction to many disruptions that bacterial cells may face.
To Walker and Gruber’s surprise, their results also showed that the total contribution of incomplete BER to cell death was different between the two proteins — Dxr-depleted cells died faster than DapB-depleted cells, suggesting that a lack of Dxr played a larger role in cell death. Because the responses to protein depletion were so different between DapB and Dxr, the researchers concluded that there is no singular pathway that causes oxidative stress; rather, it is probably a common consequence of many possible disruptions to bacterial cell physiology.
“If there’s one important thing I think we need to realize about cell death,” Gruber says, “it’s that a lot is happening to a stressed cell. And what is actually lethal might differ between two cells.”
This study adds to a body of research by Gruber, Walker, and others about the role of incomplete BER in the process of cell death. In 2012, the Walker lab published a paper in Science — building on earlier work from MIT’s Termeer Professor of Bioengineering, Jim Collins — which showed for the first time that some commonly-used antibiotics kill by way of oxidative stress and the 8-oxo-dG pathway of incomplete BER. The idea was not immediately accepted by the scientific community, and a debate ensued: Shortly after Walker’s paper, Northeastern University biologist Kim Lewis and University of Illinois biologist Jim Imlay each published separate papers suggesting that bactericidal antibiotics had nothing to do with oxidative stress. Since then, the Walker and Collins labs have continued to research the topic, producing more supporting data for their argument that oxidative stress and incomplete BER are, in fact, an important pathway of cell death.
“This new work provides a strong genetic foundation for the role of incomplete BER in bacterial cell death,” Collins says . “Oxidative stress and BER should be targeted as a means to potentiate existing antibiotics and enhance our antibiotic arsenal.”
Scientific debates like the one surrounding the contribution of incomplete BER to bacterial death are crucial to the creation of effective antibiotics. Most antibiotics work by breaking the cell wall and causing cell death that way. However, the lab’s findings offer a possibility for antibiotic assistance: the common practice of using secondary antibiotics to aid in cell death thorough a different pathway. For example, administering a secondary antibiotic that triggers the 8-oxo-dG pathway along with the primary antibiotic that is lethal to bacteria through cell wall destruction could be more effective than one antibiotic on its own, Gruber suggests.
“Many of our antibiotics are not working, or we’ve overused them in some cases, so we’re really running out of drugs,” he says. “So an antibiotic that induces oxidative stress could be another way to help existing drugs work better.”
Top image: E. coli cells with either DapB (left) or Dxr (right) depleted. Living cells are stained green while dead cells are stained red. Credit: Charley Gruber
Citation:
“Degradation of the Escherichia coli Essential Proteins DapB and Dxr Results in Oxidative Stress, which Contributes to Lethality through Incomplete Base Excision Repair”
mBio, online February 8, 2022, DOI: 10.1128/mbio.03756-21
Charley C. Gruber, Vignesh M. P. Babu, Kamren Livingston, Heer Joisher, and Graham C. Walker
December 2, 2021

Senior Desmond Edwards has an insatiable curiosity about how the human body works — and how diseases stop it from working.
Leah Campbell | School of Science
November 21, 2021
Desmond Edwards was a little kid when first learned about typhoid fever. Fortunately, he didn’t have the disease. He was looking at a cartoon public health announcement. The cartoon, produced by the Pan American Health Organization, was designed to educate people in his home country of Jamaica about the importance of immunizations for diseases like typhoid. The typhoid character in the cartoon was so unpleasant it gave him nightmares.
Edwards did have his fair share of hospital visits throughout his childhood. But, his own struggles with infection and illness, and those typhoid cartoon nightmares, became his inspiration for pursuing a career studying human disease. At age 6, Edwards was running impromptu baking soda experiments in repurposed glitter containers in his kitchen. Today, he is a senior at MIT, majoring in biology and biological engineering, thanks to a team of dedicated mentors and an insatiable curiosity about how the human body works — or, more accurately, how diseases stop it from working.
Finding a way into research
Edwards knew he wanted to do research but says he assumed that that was something you did after you got your degree. Imagine his surprise, then, upon arriving at MIT in 2018 and meeting classmates who not only had done research, but already had publications. Realizing that he could get a jump-start on his career, he sought out research opportunities and enrolled in the biology class 7.102 (Introduction to Molecular Biology Techniques) for his first-year Independent Activities Period. The class was specifically geared toward first-year students like him with no lab experience.
“It was a great first look at how research is done,” Edwards says of the class. Students took water samples from the Charles River and were expected to identify the strains of bacteria found in those samples using various biological techniques. They looked at the bacteria under a microscope. They examined how the samples metabolized different sources of carbon and determined if they could be stained by different dyes. They even got to try out basic genetic sequencing. “We knew where we were starting. And we knew the end goal,” says Edwards. The in-between was up to them.
Class 7.102 is taught by Mandana Sassanfar, a lecturer in biology and the department’s director of diversity and science outreach. For Sassanfar, the class is also an opportunity to find lab placements for students. In Edwards’ case, she literally led him to the lab of Assistant Professor Becky Lamason, walking up with him one evening to meet a postdoc, Jon McGinn, to talk about the lab and opportunities there. After Edwards expressed his interest to Lamason, she responded within 30 minutes. McGinn even followed up to answer any lingering questions.
“I think that was really what pushed it over the edge,” he says of his decision to take a position in the Lamason lab. “I saw that they were interested not only in having me as someone to help them do research, but also interested in my personal development.”
At the edges of cells and disciplines
The Lamason lab researches the life cycle of two different pathogens, trying to understand how the bacteria move between cells. Edwards has focused on Rickettsia parkeri, a tick-borne pathogen that’s responsible for causing spotted fever. This type of Rickettsia is what biologists call an obligate intracellular pathogen, meaning that it resides within cells and can only survive when it’s in a host. “I like to call it a glorified virus,” Edwards jokes.
Edwards gets excited describing the various ways in which R. parkeri can outsmart its infected host. It’s evolved to escape the phagosome of the cell, the small liquid sac that forms from the cell membrane and engulfs organisms like bacteria that pose a threat. Once it gets past the phagosome and enters the cell, it takes over cellular machinery, just like a virus. At this point of the life cycle, a bacterium will typically replicate so many times that the infected cell will burst, and the pathogen will spread widely. R. parkeri, though, can also spread to uninfected cells directly through the membrane where two cells touch. By not causing a cell to burst, the bacterium can spread without alerting the host to its presence.
“From a disease standpoint, that’s extremely interesting,” says Edwards. “If you’re not leaving the cell or being detected, you don’t see antibodies. You don’t see immune cells. It’s very hard to get that standard immune response.”
In his time in the lab, Edwards has worked on various projects related to Rickettsia, including developing genetic tools to study the pathogen and examining the potential genes that might be important in its life cycle. His projects sit at the intersection of biology and biological engineering.
“For me, I kind of live in between those spaces,” Edwards explains. “I am extremely interested in understanding the mechanisms that underlie all of biology. But I don’t only want to understand those systems. I also want to engineer them and apply them in ways that can be beneficial to society.”
Science for society
Last year, Edwards won the Whitehead Prize from the Department of Biology, recognizing students with “outstanding promise for a career in biological research.” But his extracurricular activities have been driven more by his desire to apply science for tangible social benefits.
“How do you take the science that you’ve done in the lab, in different research contexts, and translate that in a way that the public will actually benefit from it?” he asks.
Science education is particularly important for Edwards, given the educational opportunities he was given to help get to MIT. As a high schooler, Edwards participated in a Caribbean Science Foundation initiative called the Student Programme for Innovation in Science and Engineering. SPISE, as it’s known, is designed to encourage and support Caribbean students interested in careers in STEM fields. The program is modeled on the Minority Introduction to Engineering and Science program (MITES) at MIT. Cardinal Warde, a professor of electrical engineering, is himself from the Caribbean and serves as the faculty director for both MITES and SPISE.
“That experience not only kind of opened my eyes a bit more to what was available, what was in the realm of possibilities, but also provided support to get to MIT,” Edwards says of SPISE. For example, the program helped with college applications and worked with him to secure an internship at a biotech company when he first moved to the United States.
“If education falters, then you don’t replenish the field of science,” Edwards argues. “You don’t get younger generations excited, and the public won’t care.”
Edwards has also taken a leadership role in the MIT Biotechnology Group, a campus-wide student group meant to build connections between the MIT community and thought leaders in industry, business, and academia. For Edwards, the biotech and pharmaceutical industries play a clear role in disease treatment, and he knew he wanted to join the group before he even arrived at MIT. In 2019, he became co-director of the Biotech Group’s Industry Initiative, a program focused on preparing members for industry careers. In 2020, he became undergraduate president, and this year he’s co-president of the entire organization. Edwards speaks proudly of what the Biotech Group has accomplished during his tenure on the executive board, highlighting that they not only have the largest cohort ever this year, but it’s also the first time the group has been majority undergraduate.
Somehow, in between his research and outreach work, Edwards finds time to minor in French, play for the Quidditch team, and serve as co-president on the Course 20 Undergraduate Board, among other activities. It’s a balancing act that Edwards has mastered over his time at MIT because of his genuine excitement and interest in everything that he does.
“I don’t like not understanding things,” he jokes. “That applies to science, but it also extends to people.”
November 3, 2021

The PhD candidate studies the human microbiome and its proteins, while also championing the Latinx community on MIT’s campus.
Hannah Meiseles | MIT News Office
September 28, 2021
Raised in Tampa, Florida, Lindsey Backman takes pride in her family’s history and its role in the vibrant Cuban American community there. She remembers the weekends she would spend as a kid, getting café con leche with her grandparents and dancing in the studio with her friends. The cultural experiences she shared with friends, family, and neighbors growing up helped her feel comfortable being herself while growing up, and showed her from an early age how valuable a welcoming community could be to a person’s success.
Backman went on to pursue her BS in chemistry at the University of Florida. Surrounded by a diverse community, she felt supported as she leaned deeper into her interest in science. She was soon nominated to a program that matched students from underrepresented backgrounds in STEM with a university professor to pursue a summer research project. Although Backman was still uncertain about going to another university to do lab research, with encouragement from her department she gave the program a chance.
Backman matched with Professor Catherine Drennan at MIT to work on visualizing structural biology and took part in the MIT Summer Research Program in Biology (MSRP-Bio). The research clicked with her immediately and became a turning point; Backman returned to participate in the lab the following summer and then applied to graduate school.
“Getting nominated to the program changed my life. I certainly wouldn’t have applied to MIT otherwise,” says Backman. “At first, I was convinced I wouldn’t fit in, but soon found myself surrounded by people as passionate about science as I was. I knew I was in the right place.”
Uncovering secrets about the human microbiome
Today, Backman is a graduate student in the Drennan lab and researches the chemistry of the human microbiome, a collection of gut microbes essential to sustaining the body. Backman is interested in how certain bacteria can outcompete other strains by producing unique proteins that process abundant nutrients or repair broken enzymes. Her use of X-ray crystallography has helped her produce atomic models that shed light on the structure of these proteins.
One type of protein Backman and her team have characterized is called a spare part protein. When produced, this protein can help restore a broken enzyme’s ability to catalyze essential reactions. “When fixing a car with a flat tire, you would replace the tire and not the whole car. A similar strategy is being used here. These spare-part proteins act to bind and restore the activity of the enzyme completely,” she says.
Over the years, Backman has seen the depth of questions surrounding the microbiome grow. Scientists have begun to recognize how important the microbiome is to human health. “Ever since my first summer research experience at MIT, I’ve been dedicated to studying this one unique repair mechanism,” says Backman. “We’ve gone from solving the structure of the proteins to now understanding how the mechanism works. But there’s still so much more to learn — we have started to suspect these repair mechanisms speak to a broader motif in other enzymes as well.”
Backman and her team have also been leaders in characterizing how an important enzyme, called hydroxy-L-proline dehydratase (HypD), performs its unusual chemistry. This abundant enzyme takes hydroxyproline, a common nutrient in the gut, and can obtain a competitive advantage by using it as a nutrient and source of energy.
“Only a unique subset of bacteria can process hydroxyproline. On the clinical side, we have seen during infection that virulent bacteria with this ability, such as C. difficile, will start rapidly consuming hydroxyproline to proliferate,” says Backman. “Conversely, we could one day create antibiotics that specifically inhibit HypD without killing our beneficial bacteria.”
Encouraging the future of science
Outside of her research, Backman cares deeply about serving and being a part of the Latinx community on campus. She helped co-found the MIT Latinx Graduate Student Association and has served for four years as a graduate resident assistant for La Casa, the Latinx undergraduate living community at New House. “La Casa is a really tight-knit and familial community,” says Backman. “Some of our original freshmen are now seniors, so it’s been really rewarding to see their whole transition throughout college. I love getting to watch students explore and come to realize what they’re passionate about.”
Backman has also been instrumental in spurring equity initiatives on campus. She is currently a student representative for the MIT Department of Chemistry Diversity, Equity, and Inclusion Committee and has worked to implement programs that support the success of underrepresented groups on campus. Her five years of service as an MIT Chemistry Access Program mentor have encouraged many underrepresented undergraduate students to pursue chemistry graduate programs. For all her hard work at improving MIT’s campus, Backman recently received the Hugh Hampton Young Fellowship.
In the future, Backman aspires to continue researching the microbiome and mentoring students by becoming a professor. She hopes to continue the cycle and inspire more young scientists to recognize their inner potential. “I was never one of those kids that knew I wanted to be a scientist someday. My PI completely changed my life, and I would not be at MIT today without her,” she says. “Having mentors that believe in you at critical points in your life can make all the difference.”
“I think there’s this wrong assumption that diversity initiative work takes away from time that could be spent doing science. In my mind, we need to recognize how these things go hand in hand,” says Backman.
“The only way we’re going to get the best scientists is by creating a healthier, more diverse environment where people of all backgrounds feel welcomed. It’s only when people feel comfortable that they can make their greatest contributions to the field.”

Bailey Bowcutt investigated COVID-19 cases in rural Wyoming before coming to MIT for the summer and applying her knowledge to a new cellular invader.
Raleigh McElvery
July 23, 2021
The first time Bailey Bowcutt saw a lab it was nothing like she expected. Rather than a stark, sterile setting with sullen figures floating around like ghosts in white lab coats, the atmosphere was cordial and the dress casual. Some scientists even sported vibrant shirts with Marvel characters. A high school senior on a class field trip, Bowcutt couldn’t have predicted that the next time she’d set foot in the Wyoming Public Health Laboratory she’d no longer be a visitor, but a researcher performing diagnostic testing during a global pandemic. Now, as COVID-19 restrictions begin to lift, she’s taking the research tools she’s learned to Cambridge, Massachusetts to complete the Bernard S. and Sophie G. Gould MIT Summer Research Program in Biology (BSG-MSRP-Bio) and investigate how other types of pathogens spread.
Growing up in rural Wyoming, Bowcutt had little exposure to science because there were few research institutes close by. But watching family members suffer from gastrointestinal illness and other infections spurred her to pursue a degree in microbiology at Michigan State University (MSU). Shortly after she arrived on campus in the fall of 2019, she joined Shannon Manning’s lab studying antibiotic resistance in cattle.
Cows are prone to contracting a bacterial infection of the udder called mastitis. (In humans, a similar inflammation can occur in breast tissue.) Manning’s lab is looking at how antibiotic treatments affect the bovine gut microbiome and emergence of antibiotic resistance genes. Bowcutt’s role was to help identify these super bugs inside the cows’ gastrointestinal tracts.
“I got to go to the farm to take samples, which involved a glove that goes all the way up to the shoulder and some invasive maneuvers inside cows,” she explains. “Luckily, I was just the bag holder!”
Intimate sample collection aside, Bowcutt was excited about the work because it combined agriculture and human health research to solve issues plaguing rural communities. But her time on the farm was cut short when COVID-19 cases climbed in early 2020. She headed back to her home in Wyoming to begin remote MSU classes and, reminiscing about her field trip to the Wyoming Public Health Laboratory, reached out to the director to see if there were any internship opportunities.
“I’d barely learned how to do science at that point, but they needed people who could handle a pipette, so they took me,” she says. “I ended up being one of the first people there helping with COVID research, and I stayed for about a year-and-a-half while I took online classes.”
The lab would receive nasopharyngeal swabs from COVID-19 patients, and Bowcutt’s first task was to help extract RNA from the samples. Later, she transitioned to another project, which required performing PCR on untreated wastewater samples to glean a population-level understanding of where COVID-19 outbreaks were occurring.
She began toying with the idea of pursuing a PhD, but wasn’t sure what it would entail. So, in early 2021, she started Googling summer science programs and stumbled on BSG-MSRP-Bio. She was accepted, and paired with one of the very labs that had caught her eye online: assistant professor Becky Lamason’s group.
“If you’ve ever seen microscopy pictures from the Lamason lab, they’re just so beautiful,” Bowcutt explains. Beautiful, yes — but she would soon learn these snapshots capture a chilling cellular invasion and molecular heist.
The Lamason lab watches malicious bacteria as they hijack molecules in human host cells to build long tails, rocket around, and punch through the cell membrane to spread. Bowcutt’s mentor, graduate student Yamilex Acevedo-Sánchez, focuses on the food-borne bacterium Listeria monocytogenes, which targets the gastrointestinal tract. Acevedo-Sánchez’s research aims to understand the host cell pathways that Listeria commandeers to move from one cell to the next in a process called cell-to-cell spread.
Together, Acevedo-Sánchez and Bowcutt are investigating several proteins in the human host cell involved in cellular transport and membrane remodeling (Caveolin-1, Pacsin2, and Fes), which could regulate Listeria’s spread. Over the summer, the duo has been adjusting the levels of these proteins and observing what happens to Listeria’s ability to move from cell-to-cell.
Bowcutt spends most of her days doing Western blots; growing Listeria and mammalian cells; and combining immunofluorescence assays with fixed and live cell microscopy to take her own striking microscopy images and movies of the parasites.
“I expected the work environment at MIT to be very intense, but everyone has been really friendly and willing to answer questions,” she says. “Some of my favorite experiences have just been in the lab while everyone is bustling around. It’s a welcome change after so much COVID-19 isolation.”
Now that the COVID-era occupancy restrictions have lifted, Bowcutt’s lab bench neighbor is Lamason herself. “She’s next to me doing experiments all the time,” Bowcutt explains, “which is cool because she’s really engaging with the research in the same way we are.”
Bowcutt says her summer experience has given her some much-needed practice designing research questions and devising the experiments to answer them. She’s also acquired a new skill she didn’t anticipate: interpreting ambiguous results and developing follow-up experiments to clarify them.
These days, the prospect of a PhD seems much less intimidating. In fact, the Lamason lab has done more than simply pique Bowcutt’s interested in fundamental biology research. She’s now considering ways to combine her microbiology skills with her interest in rural health care.
“I didn’t expect to see this much growth in myself,” she says, “and I know it’s making me a better scientist. I’m excited to return to MSU in the fall because I feel like I can do so much more now — and I would totally do it again.”