Small changes in the molecular machines that carry out RNA interference can lead to big differences in the efficacy of gene silencing. These new findings from the Bartel Lab have implications for the design of gene-silencing therapeutics.
Greta Friar | Whitehead Institute
July 17, 2024
RNA interference (RNAi) is a process that many organisms, including humans, use to decrease the activity of target RNAs in cells by triggering their degradation or slicing them in half. If the target is a messenger RNA, the intermediary between gene and protein, then RNAi can decrease or completely silence expression of the gene. Researchers figured out how to tailor RNAi to target different RNAs, and since then it has been used as a research tool to silence genes of interest. RNAi is also used in a growing number of therapeutics to silence genes that contribute to disease.
However, researchers still do not understand some of the biochemistry underlying RNAi. Slight differences in the design of the RNAi machinery can lead to big differences in how effective it is at decreasing gene expression. Through trial and error, researchers have worked out guidelines for making the most effective RNAi tools without understanding exactly why they work. However, Whitehead Institute Member David Bartel and graduate student in his lab Peter Wang have now dug deeper to figure out the mechanics of the main cellular machine involved in RNAi. The researchers’ findings, shared in Molecular Cell on July 17, not only provide explanations for some of the known rules for RNAi tool design, but also provide new insights that could improve future designs.
Slicing speed is highly variable
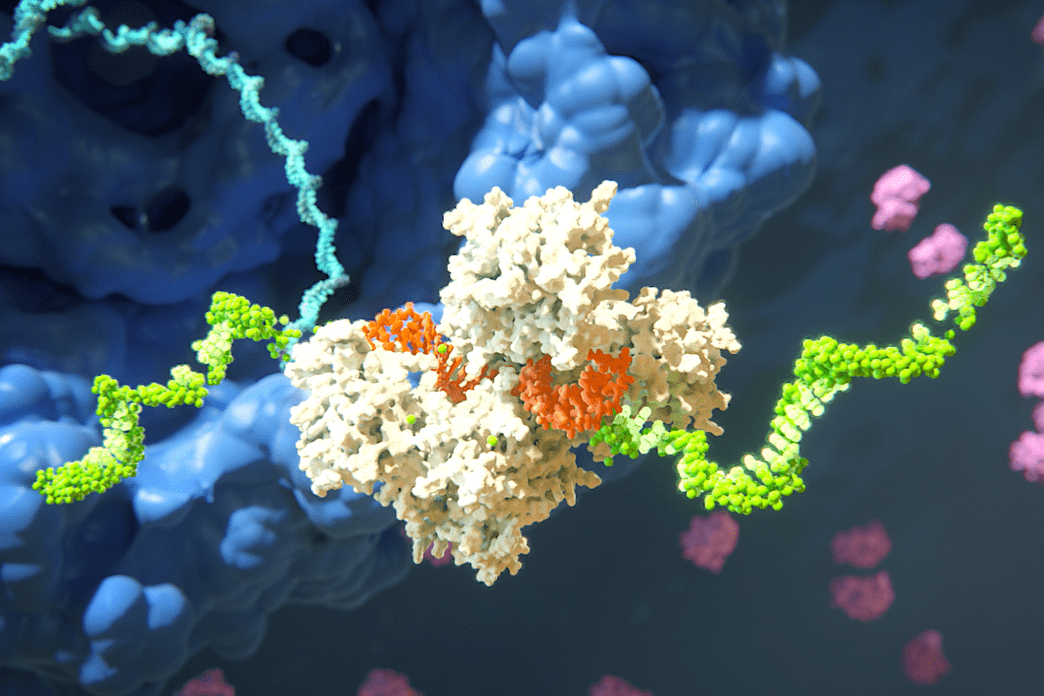
The cellular machine that carries out RNAi has two main parts. One is a guide RNA, a tiny RNA typically only 22 bases or nucleotides long. RNA, like DNA, is made of four possible bases, although RNA has the base uracil (U) instead of the DNA base thymine (T). RNA bases bind to each other in certain pairings—guanines (G) pair to cytosines (C) and adenines (A) pair to U’s—and the sequence of bases in the guide RNA corresponds to a complementary sequence within the target RNA. When the guide RNA comes across a target, the corresponding bases pair up, binding the RNAs. Then the other part of the RNAi machine, an Argonaute protein bound to the guide RNA, can slice the target RNA in half or trigger the cell to break it down more gradually.
In humans, AGO2 is the Argonaute protein that is best at slicing. Only a couple dozen RNA targets actually get sliced, but these few targets play essential roles in processes such as neuron signal control and accurate body shape formation. Slicing is also important for RNAi tools and therapeutics.
In order for AGO2 to slice its target, the target must be in the exact right position. As the guide and target RNAs bind together, they go through a series of motions to ultimately form a double helix. Only in that configuration can AGO2 slice the target.
Researchers had assumed that AGO2 slices through different target RNAs at roughly the same rate, because most research into this process used the same few guide RNAs. These guide RNAs happen to have similar features, and so similar slicing kinetics—but they turn out not to be representative of most guide RNAs.
Wang paired AGO2 with a larger variety of guide RNAs and measured the rate at which each AGO2-guide RNA complex sliced its targets. He found big differences. Whereas the commonly used guide RNAs might differ in their slicing rate by 2-fold, the larger pool of guide RNAs differed by as much as 250-fold. The slicing rates were often much slower than the researchers expected. Previously, researchers thought that all targets could be sliced relatively quickly, so the rate wasn’t considered as a limiting factor – other parts of the process were thought to determine the overall pace – but Wang found that slicing can sometimes be the slowest step.
“The important consideration is whether the slicing rate is faster or slower than other processes in the cell,” Wang says. “We found that for many guide RNAs, the slicing rate was the limiting factor. As such, it impacted the efficacy.”
The slower AGO2 is to slice targets, the more messenger RNAs will remain intact to be made into protein, meaning that the corresponding gene will continue being expressed. The researchers observed this in action: the guide RNAs with slower slicing rates decreased target gene expression by less than the faster ones.
Small changes lead to big differences in slicing rate
Next, the researchers explored what could be causing such big differences in slicing rate between guide RNAs. They mutated guide RNAs to swap out single bases along the guide RNA’s sequence—say, switching the 10th base in the sequence from a C to an A—and measured how this changed the slicing rate.
“The important consideration is whether the slicing rate is faster or slower than other processes in the cell,” Wang says. “We found that for many guide RNAs, the slicing rate was the limiting factor. As such, it impacted the efficacy.”
The researchers found that slicing rate increased when the base at position 7 was an A or a U. The bases A and U pair more weakly than C and G. The researchers found that having a weak A-U pair at that position, or a fully mismatched pair at position 6 or 7, may allow a kink to form in the double helix shape that actually makes the target easier to slice. Wang also found that slicing rate increases with certain substitutions at the 10th and the 17th base positions, although the researchers could not yet determine possible underlying mechanisms.
These observations correspond to existing recommendations for RNAi design, such as not using a G at position 7. The new work demonstrates that the reason these recommendations work is because they affect the slicing rate, and, in the case of position 7, the new work further identifies the specific mechanism at play.
Interplay between regions matters
People designing synthetic guide RNAs thought that the bases at the tail end, past the 16th position, were not very important. This is because in the case of the most commonly used guide RNAs, the target will be rapidly cleaved even if all of the tail end positions are mismatches that cannot pair.
However, Wang and Bartel found that the identity of the tail end bases are only irrelevant in a specific scenario that happens to be true of the most commonly used guide RNAs: when the bases in the center of the guide RNA (positions 9-12) are strong-pairing Cs and Gs. When the center pairings are weak, then the tail end bases need to be perfect matches to the target RNA. The researchers found that guide RNAs could have up to a 600-fold difference in tolerance for tail end mismatches based on the strength of their central pairings.
The reason for this difference has to do with the final set of motions that the two RNAs must perform in order to assume their final double helix shape. A perfectly paired tail end makes it easier for the RNAs to complete these motions. However, a strong enough center can pull the RNAs into the double helix even if the tail ends are not ideally suited for doing so.
The observation that weak central pairing requires perfect or near perfect tail end matches could provide a useful new guideline for designing synthetic RNAs. Any guide RNA runs the risk of sometimes binding other messenger RNAs that are similar enough to the intended target RNA. In the case of a therapy, this off-target binding can lead to negative side effects. Bartel and Wang suggest that researchers could design guide RNAs with weak centers, which would require more perfect pairing in the tail end, so that the guide RNA will be less likely to bind non-target RNAs; only the perfect pairing of the target’s RNA sequence would suffice.
Altogether, Wang and Bartel’s findings explain how small differences between guide RNAs can make such large differences in the efficacy of RNAi, providing a rationale for the long-standing RNAi design guidelines. Some of the findings even suggest new guidelines that could help with future synthetic guide RNA designs.
“Discovering the interplay between the center and tail end of the guide RNA was unexpected and satisfying,” says Bartel, who is also a professor at the Massachusetts Institute of Technology and a Howard Hughes Medical Investigator. “It explains why, even though the guidelines suggested that tail-end sequence doesn’t matter, the target RNAs that are sliced in our cells do have pairing to the tail end. This observation could prove useful to reduce off-target effects in RNAi therapeutics.”