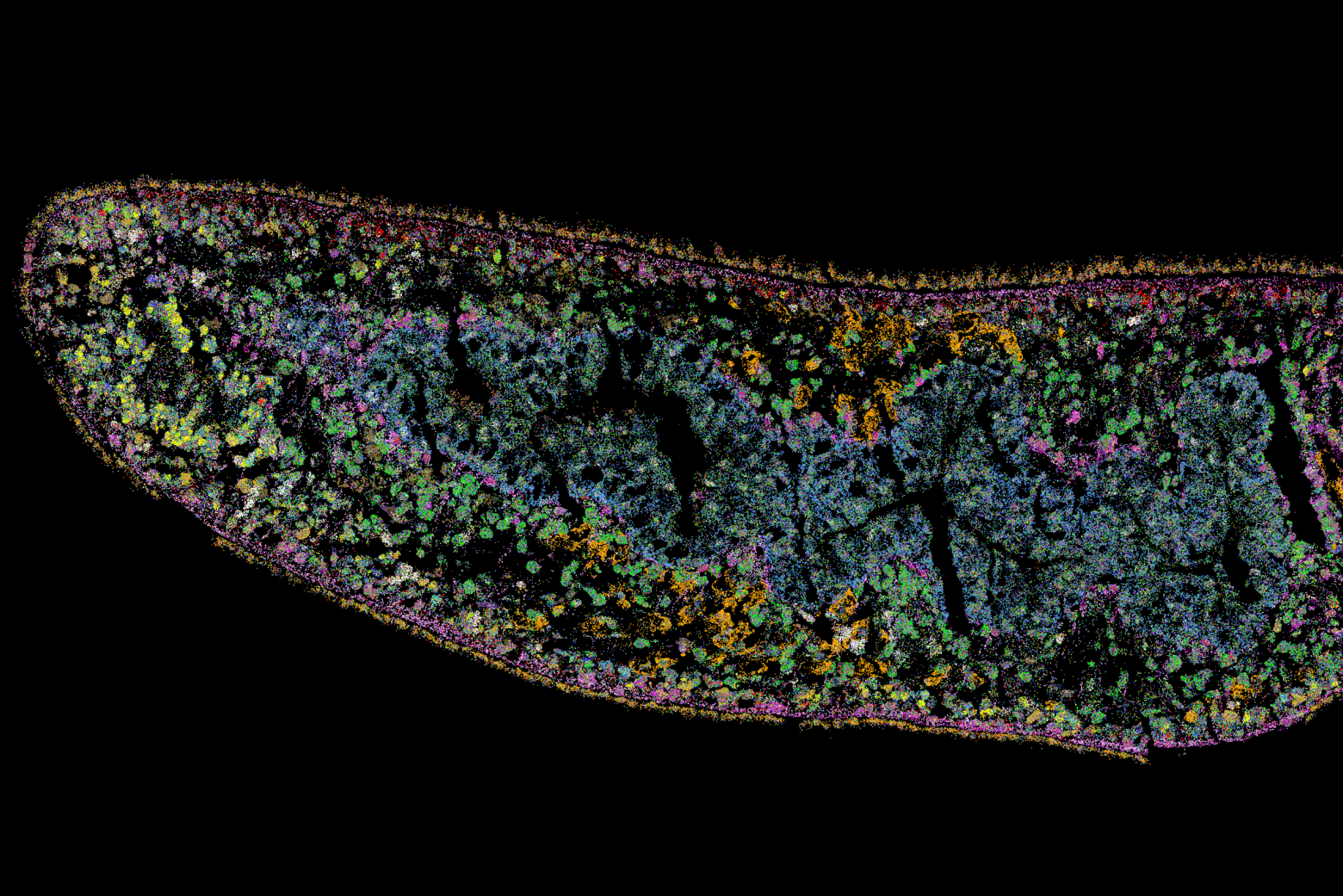
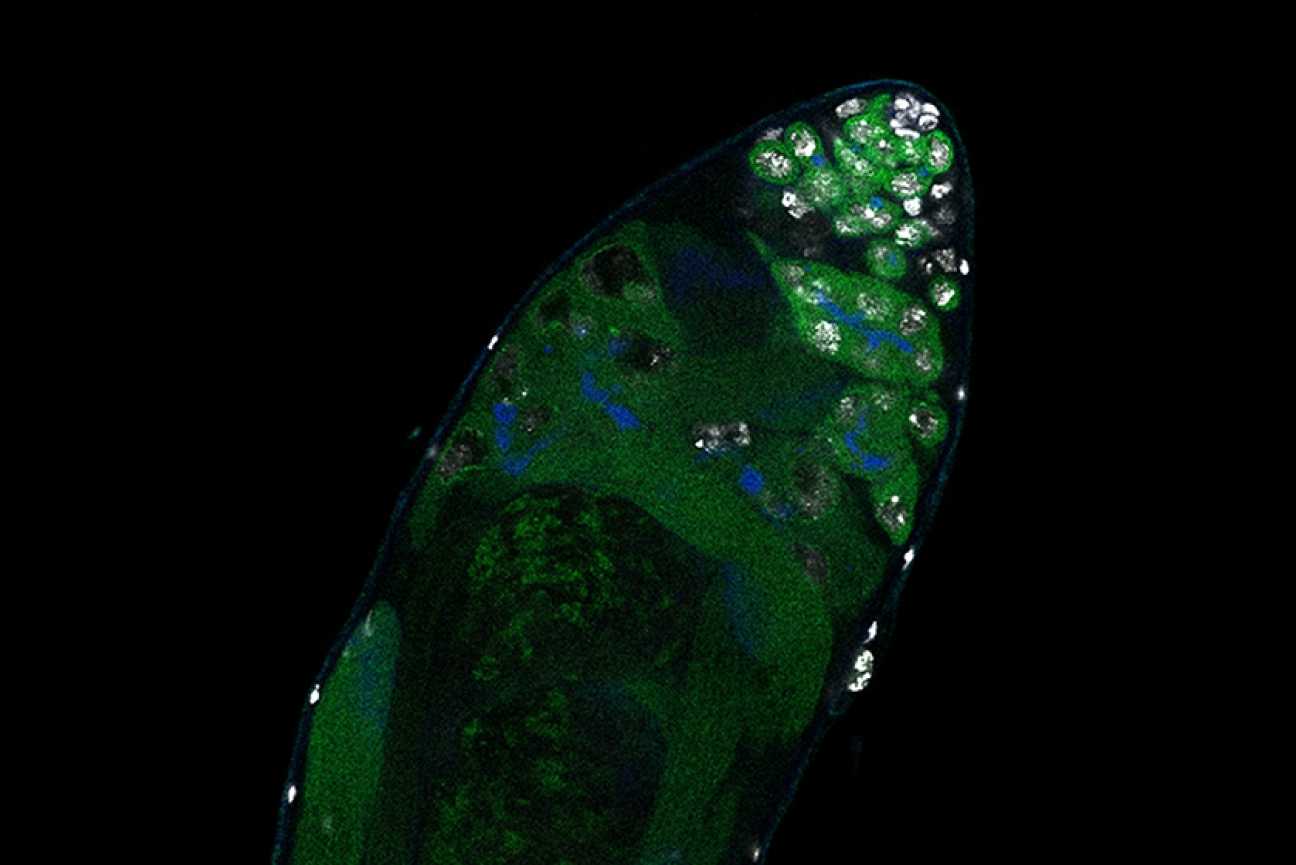
Germline stem cells are the pool of stem cells capable of becoming eggs or sperm. They divide asymmetrically, such that one of the cells resulting from a division is another stem cell and the other is a differentiated cell, which has progressed one step further down the path towards becoming an egg or sperm. Researchers have thought that this asymmetrical division served to replenish the pool of stem cells—making sperm or eggs, but also making more stem cells to produce future sperm or eggs. However, the germline has another way to replenish itself: cells that have differentiated only one or two steps down the path to becoming eggs or sperm are capable of reverting into stem cells. Why, then, do stem cells divide asymmetrically?
New research from Whitehead Institute Member Yukiko Yamashita, who is also a professor of biology at the Massachusetts Institute of Technology and an HHMI Investigator, and former postdoc in her lab Jonathan Nelson shows that asymmetrical division in germline stem cells serves a different but equally important purpose in male fruit flies (Drosophila melanogaster), a common model animal for germline research. The work, published in the journal Proceedings of the National Academy of Sciences (PNAS) on November 13, suggests that in flies, germline stem cells divide asymmetrically in order to unequally split a certain kind of DNA, called ribosomal DNA (rDNA), between the two dividing cells and then keep the cell with more rDNA in the stem cell pool. This is necessary in order to keep the germline viable over generations of cell divisions, and so to keep individual flies fertile and capable of reproduction. The researchers show that only germline stem cells, and not other types of germ cells, drive this process, and explain why stem cells’ asymmetric divisions make them uniquely suited to maintaining rDNA.
Ribosomal DNA is critical to maintain in the germline because it contains the instructions for making a major part of ribosomes, the cellular machines that build proteins from genetic instructions. Proteins are the main workhorses of the cell, and so cells need to make many ribosomes in order to build all of the proteins that they need. Consequently, rDNA exists as many copies repeated in a row of the code for components of the ribosome. All of these repeats make it easy for the cell to mass produce ribosomes, but they also come with a risk: repetitive DNA is prone to losing repeats during cell division. When the cell’s rDNA is copied, it’s easy for a few of the many identical repeats to get cut out, so that the resulting copy of the genome has fewer rDNA repeats than the original.
Most cells can afford to lose a few rDNA repeats without too many negative effects, but the germline cannot. Whereas other cells die with the body they are in, germ cells produce eggs and sperm that will form a new body, which produces new germ cells, and so on. The germ cell lineage is effectively immortal. Over the course of its endless cycle of cell division, the loss of rDNA repeats would add up until the cells became dysfunctional and then died. This would make the individual bearing those germ cells infertile, and so cause their lineage to go extinct.
Researchers have known that germ cells have some way to regain rDNA repeats when the number gets too low—if germ cells couldn’t do this, none of us would exist—but the details of how cells achieve this have been largely mysterious. One proposed model was that when a germ cell divides, sometimes it might divide up its rDNA unequally between the two resulting cells, so that one cell would gain rDNA repeats. Yamashita and Nelson have previously found evidence that this model is correct, and they discovered some of the specific mechanisms that enable it to happen. In a 2023 PNAS paper, the researchers showed that a retrotransposon, a “selfish” genetic element whose function is to make more copies of itself, actually helps germ cells maintain rDNA. During cell division, the retrotransposon R2 slices open one copy of the chromosome containing rDNA in its quest to insert extra copies of itself into the genome. The cell tries to repair the break using the copy on the other intact chromosome, but the tricky nature of repetitive DNA can cause the cell to lose its place, so that it stitches a stretch of rDNA repeats from one copy of the chromosome into the other copy instead.
Through this process, the germline can boost the level of rDNA in a cell—but only by as much as another cell loses. How does this win-lose exchange lead to an overall increase in rDNA levels across the germline cell population to compensate for lost rDNA? In this latest work, Yamashita and Nelson show through mathematical modeling that in cells that divide symmetrically, it would not. Gains and losses in rDNA through this form of exchange would occur essentially at random and cancel each other out over time.
Now consider an asymmetric division. After a germline stem cell divides, the cell that differentiates will go through a few more divisions and ultimately create a specific number of sperm cells–the number happens to be sixty-four. If this daughter cell gets the chromosome with more rDNA repeats, then that would lead to sixty-four sperm with more rDNA repeats—but that would be it, as the sperm have exited the pool of replicating germline stem cells.
However, the daughter cell that remains a germline stem cell will divide again to create a differentiated cell (which will become sixty-four sperm) and another stem cell, which will divide again, leading to another sixty-four sperm and another stem cell—and so on. All of these cells, including many sperm, would inherit the higher number of rDNA repeats. Furthermore, at each division, there would be an opportunity for another unequal split of rDNA. As long as the stem cell always gets the boost in rDNA, then the cumulative number of rDNA repeats would keep growing in the overall population over time—and Yamashita and colleagues’ past work shows that the germline can ensure this. A 2022 Science Advances paper from Yamashita and then-postdoc in her lab George Watase showed that when a germline stem cell divides, the DNA strand with more rDNA repeats is tagged with a protein that the researchers named Indra, which helps mark it to stay in the daughter cell that will become another stem cell. Yamashita and Nelson’s new paper includes mathematical modeling by second author Tomohiro Kumon, a postdoc in Yamashita’s lab, that proves that this is not only sufficient to restore the level of rDNA repeats over time, but that it is the most effective and efficient way for the germline to do so.
“There was this problem with the unequal exchange model of rescuing rDNA, because every cell that gained rDNA did so at the expense of another that was losing it,” Nelson says. “What we show here is that the reason why there’s a bias towards gain in the germline is because this process is happening within these asymmetrically dividing germline stem cells that can gain and gain and gain, while the cells that lose rDNA exit the cycle and so have a limited effect.”
The researchers complemented their mathematical modeling with evidence that the process to increase rDNA repeats occurs primarily or solely in germline stem cells. They found that when the number of rDNA repeats got low enough, then expression of R2 and the presence of double-stranded DNA breaks both increased in germline stem cells, but not significantly in other germ cell types.
Yamashita and Nelson propose that the different cell types in the germline take on different functions to create a pipeline for maximizing the health of future sperm. Germ cells that are one or two steps down the path of differentiation from stem cells are essentially identical to them, to the point that they can be difficult to tell apart in testing, but they divide symmetrically. They are also much more sensitive to DNA damage; the researchers found that R2 exposure kills these cells.
Germline stem cells, with their asymmetrical division and ability to tolerate R2 expression, serve to restore rDNA levels when they get too low. Then the differentiated germ cells serve to weed out mutations—including those introduced during R2 expression in the earlier stem cell stage—by killing off cells with DNA damage. The different strengths of the different types of germ cells creates an effective pipeline to produce the largest number of sperm cells with high rDNA repeat number and low DNA damage.
Eventually, this new understanding of the details of how cells maintain their rDNA could lead to medical therapies. For example, cancer cells are, like germ cells, an essentially immortal cell line, and so must have a way to maintain their rDNA. If researchers could someday find a way to prevent them from doing so, that could be a good treatment strategy. The work also may have implications for research on aging, as rDNA decreases with age in other cell types. In the meantime, Yamashita and Nelson are excited to have solved several long-standing mysteries in their field, including how germ cells can restore rDNA at a population level when each division creates an equal loss and gain of rDNA, and why germline stem cells divide asymmetrically.
“Typically, when you publish a paper, you feel like you’ve fit two puzzle pieces together, but in this case, I feel like we fit a bunch of puzzle pieces together,” Yamashita says. “It’s been immensely satisfying to find answers to multiple questions and see how they all fit together to explain the mechanisms of this process that’s necessary for germline immortality.”

Sipping a beer on a warm summer evening, one might not consider that humans and yeast have been inextricably linked for thousands of years; winemaking, baking, and brewing all depend on budding yeast. Outside of baking and fermentation, researchers also use Saccharomyces cerevisiae, classified as a fungus, to study fundamental questions of cell biology.
Budding yeast gets its name from the way it multiplies. A daughter cell forms first as a swelling, protruding growth on the mother cell. The daughter cell projects further and further from the mother cell until it detaches as an independent yeast cell.
How do cells decide on a front and back? How do cells decode concentration gradients of chemical signals to orient in useful directions, or sense and navigate around physical obstacles? New Department of Biology faculty member Daniel “Danny” Lew uses the model yeast S. cerevisiae, and a non-model yeast with an unusual pattern of cell division, to explore these questions.
Q: Why is it useful to study yeast, and how do you approach the questions you hope to answer?
A: Humans and yeast are descended from a common ancestor, and some molecular mechanisms developed by that ancestor have been around for so long that yeast and mammals often use the same mechanisms. Many cells develop a front and migrate or grow in a particular direction, like the axons in our nervous system, using similar molecular mechanisms to those of yeast cells orienting growth towards the bud.
When I started my lab, I was working on cell cycle control, but I’ve always been interested in morphogenesis and the cell biology of how cells change shape and decide to do different things with different parts of themselves. Those mechanisms turn out to be conserved between yeast and humans.
But some things are very different about fungal and animal cells. One of the differences is the cell wall and what fungal cells have to do to deal with the fact that they have a cell wall.
Fungi are inflated by turgor pressure, which pushes their membranes against the rigid cell wall. This means they’ll die if there is any hole in the cell wall, which would be expected to happen often as cells remodel the wall in order to grow. We’re interested in understanding how fungi sense when any weak spots appear in the wall and repair them before those weak spots become dangerous.
Yeast cells, like most fungi, also mate by fusing with a partner. To succeed, they must do the most dangerous thing in the fungal lifecycle: get rid of the cell wall at the point of contact to allow fusion. That means they must be precise about where and when they remove the wall. We’re fascinated to understand how they know it is safe to remove the wall there, and nowhere else.
We take an interdisciplinary approach. We’ve used genetics, biochemistry, cell biology, and computational biology to try and solve problems in the past. There’s a natural progression: observation and genetic approaches tend to be the first line of attack when you know nothing about how something works. As you learn more, you need biochemical approaches and, eventually, computational approaches to understand exactly what mechanism you’re looking at.
I’m also passionate about mentoring, and I love working with trainees and getting them fascinated by the same problems that fascinate me. I’m looking to work with curious trainees who love addressing fundamental problems.
Q: How does yeast decide to orient a certain way—towards a mating partner, for example?
A: We are still working on questions of how cells analyze the surrounding environment to pick a direction. Yeast cells have receptors that sense pheromones that a mating partner releases. What is amazing about that is that these cells are incredibly small, and pheromones are released by several potential partners in the neighborhood. That means yeast cells must interpret a very confusing landscape of pheromone concentrations. It’s not apparent how they manage to orient accurately toward a single partner.
That got me interested in related questions. Suppose the cell is oriented toward something that isn’t a mating partner. The cell seems to recognize that there’s an obstacle in the way, and it can change direction to go around that obstacle. This is how fungi get so good at growing into things that look very solid, like wood, and some fungi can even penetrate Kevlar vests.
If they recognize an obstacle, they have to change directions and go around it. If they recognize a mating partner, they have to stick with that direction and allow the cell wall to get degraded. How do they know they’ve hit an obstacle? How do they know a mating partner is different from an obstacle? These are the questions we’d like to understand.
Q: For the last couple of years, you’ve also been studying a budding yeast that forms multiple buds when it reproduces instead of just one. How did you come across it, and what questions are you hoping to explore?
A: I spent several years trying to figure out why most yeasts make one bud and only one bud, which I think is related to the question of why migrating cells make one and only one front. We had what we thought was a persuasive answer to that, so seeing a yeast completely disobey that and make as many buds as it felt like was a shock, which got me intrigued.
We started working on it because my colleague,Amy Gladfelter, had sampled the waters around Woods Hole, Massachusetts. When she saw this specimen under a microscope, she immediately called me and said, “You have to look at this.”
A question we’re very intrigued by is if the cell makes five, seven, or 12 buds simultaneously, how do they divide the mother cell’s material and growth capacity five, seven, or 12 ways? It looks like all of the buds grow at the same rate and reach about the same size. One of our short-term goals is to check whether all the buds really get to exactly the same size or whether they are born unequal.
And we’re interested in more than just growth rate. What about organelles? Do you give each bud the same number of mitochondria, nuclei, peroxisomes, and vacuoles? That question will inevitably lead to follow-up questions. If each bud has the same number of mitochondria, how does the cell measure mitochondrial inheritance to do that? If they don’t have the same amount, then buds are each born with a different complement and ratio of organelles. What happens to buds if they have very different numbers of organelles?
As far as we can tell, every bud gets at least one nucleus. How the cell ensures that each bud gets a nucleus is a question we’d also very much like to understand.
We have molecular candidates because we know a lot about how model yeasts deliver nuclei, organelles, and growth materials from the mother to the single bud. We can mutate candidate genes and see if similar molecular pathways are involved in the multi-budding yeast and, if so, how they are working.
It turns out that this unconventional yeast has yet to be studied from the point of view of basic cell biology. The other thing that intrigues me is that it’s a poly-extremophile. This yeast can survive under many rather harsh conditions: it’s been isolated in Antarctica, from jet engines, from all kinds of plants, and of course from the ocean as well. An advantage of working with something so ubiquitous is we already know it’s not toxic to us under almost any circumstances. We come into contact with it all the time. If we learn enough about its cell biology to begin to manipulate it, then there are many potential applications, from human health to agriculture.

Humans split away from our closest animal relatives, chimpanzees, and formed our own branch on the evolutionary tree about seven million years ago. In the time since—brief, from an evolutionary perspective—our ancestors evolved the traits that make us human, including a much bigger brain than chimpanzees and bodies that are better suited to walking on two feet. These physical differences are underpinned by subtle changes at the level of our DNA. However, it can be hard to tell which of the many small genetic differences between us and chimps have been significant to our evolution.
New research from Whitehead Institute Member Jonathan Weissman; University of California, San Francisco Assistant Professor Alex Pollen; Weissman lab postdoc Richard She; Pollen lab graduate student Tyler Fair; and colleagues uses cutting edge tools developed in the Weissman lab to narrow in on the key differences in how humans and chimps rely on certain genes. Their findings, published in the journal Cell on June 20, may provide unique clues into how humans and chimps have evolved, including how humans became able to grow comparatively large brains.
Studying function rather than genetic code
Only a handful of genes are fundamentally different between humans and chimps; the rest of the two species’ genes are typically nearly identical. Differences between the species often come down to when and how cells use those nearly identical genes. However, only some of the many differences in gene use between the two species underlie big changes in physical traits. The researchers developed an approach to narrow in on these impactful differences.
Their approach, using stem cells derived from human and chimp skin samples, relies on a tool called CRISPR interference (CRISPRi) that Weissman’s lab developed. CRISPRi uses a modified version of the CRISPR/Cas9 gene editing system to effectively turn off individual genes. The researchers used CRISPRi to turn off each gene one at a time in a group of human stem cells and a group of chimp stem cells. Then they looked to see whether or not the cells multiplied at their normal rate. If the cells stopped multiplying as quickly or stopped altogether, then the gene that had been turned off was considered essential: a gene that the cells need to be active–producing a protein product–in order to thrive. The researchers looked for instances in which a gene was essential in one species but not the other as a way of exploring if and how there were fundamental differences in the basic ways that human and chimp cells function.
By looking for differences in how cells function with particular genes disabled, rather than looking at differences in the DNA sequence or expression of genes, the approach ignores differences that do not appear to impact cells. If a difference in gene use between species has a large, measurable effect at the level of the cell, this likely reflects a meaningful difference between the species at a larger physical scale, and so the genes identified in this way are likely to be relevant to the distinguishing features that have emerged over human and chimp evolution.
“The problem with looking at expression changes or changes in DNA sequences is that there are many of them and their functional importance is unclear,” says Weissman, who is also a professor of biology at the Massachusetts Institute of Technology and an Investigator with the Howard Hughes Medical Institute. “This approach looks at changes in how genes interact to perform key biological processes, and what we see by doing that is that, even on the short timescale of human evolution, there has been fundamental rewiring of cells.”
After the CRISPRi experiments were completed, She compiled a list of the genes that appeared to be essential in one species but not the other. Then he looked for patterns. Many of the 75 genes identified by the experiments clustered together in the same pathways, meaning the clusters were involved in the same biological processes. This is what the researchers hoped to see. Individual small changes in gene use may not have much of an effect, but when those changes accumulate in the same biological pathway or process, collectively they can cause a substantive change in the species. When the researchers’ approach identified genes that cluster in the same processes, this suggested to them that their approach had worked and that the genes were likely involved in human and chimp evolution.
“Isolating the genetic changes that made us human has been compared to searching for needles in a haystack because there are millions of genetic differences, and most are likely to have negligible effects on traits,” Pollen says. “However, we know that there are lots of small effect mutations that in aggregate may account for many species differences. This new approach allows us to study these aggregate effects, enabling us to weigh the impact of the haystack on cellular functions.”
Researchers think bigger brains may rely on genes regulating how quickly cells divide
One cluster on the list stood out to the researchers: a group of genes essential to chimps, but not to humans, that help to control the cell cycle, which regulates when and how cells decide to divide. Cell cycle regulation has long been hypothesized to play a role in the evolution of humans’ large brains. The hypothesis goes like this: Neural progenitors are the cells that will become neurons and other brain cells. Before becoming mature brain cells, neural progenitors divide multiple times to make more of themselves. The more divisions that the neural progenitors undergo, the more cells the brain will ultimately contain—and so, the bigger it will be. Researchers think that something changed during human evolution to allow neural progenitors to spend less time in a non-dividing phase of the cell cycle and transition more quickly towards division. This simple difference would lead to additional divisions, each of which could essentially double the final number of brain cells.
Consistent with the popular hypothesis that human neural progenitors may undergo more divisions, resulting in a larger brain, the researchers found that several genes that help cells to transition more quickly through the cell cycle are essential in chimp neural progenitor cells but not in human cells. When chimp neural progenitor cells lose these genes, they linger in a non-dividing phase, but when human cells lose them, they keep cycling and dividing. These findings suggest that human neural progenitors may be better able to withstand stresses—such as the loss of cell cycle genes—that would limit the number of divisions the cells undergo, enabling humans to produce enough cells to build a larger brain.
“This hypothesis has been around for a long time, and I think our study is among the first to show that there is in fact a species difference in how the cell cycle is regulated in neural progenitors,” She says. “We had no idea going in which genes our approach would highlight, and it was really exciting when we saw that one of our strongest findings matched and expanded on this existing hypothesis.”
More subjects lead to more robust results
Research comparing chimps to humans often uses samples from only one or two individuals from each species, but this study used samples from six humans and six chimps. By making sure that the patterns they observed were consistent across multiple individuals of each species, the researchers could avoid mistaking the naturally occurring genetic variation between individuals as representative of the whole species. This allowed them to be confident that the differences they identified were truly differences between species.
The researchers also compared their findings for chimps and humans to orangutans, which split from the other species earlier in our shared evolutionary history. This allowed them to figure out where on the evolutionary tree a change in gene use most likely occurred. If a gene is essential in both chimps and orangutans, then it was likely essential in the shared ancestor of all three species; it’s more likely for a particular difference to have evolved once, in a common ancestor, than to have evolved independently multiple times. If the same gene is no longer essential in humans, then its role most likely shifted after humans split from chimps. Using this system, the researchers showed that the changes in cell cycle regulation occurred during human evolution, consistent with the proposal that they contributed to the expansion of the brain in humans.
The researchers hope that their work not only improves our understanding of human and chimp evolution, but also demonstrates the strength of the CRISPRi approach for studying human evolution and other areas of human biology. Researchers in the Weissman and Pollen labs are now using the approach to better understand human diseases—looking for the subtle differences in gene use that may underlie important traits such as whether someone is at risk of developing a disease, or how they will respond to a medication. The researchers anticipate that their approach will enable them to sort through many small genetic differences between people to narrow in on impactful ones underlying traits in health and disease, just as the approach enabled them to narrow in on the evolutionary changes that helped make us human.

Whitehead Institute Member Siniša Hrvatin has been named as one of the 15 researchers to be selected as 2023 Searle Scholars. The Searle Scholars Program supports the research of exceptional young faculty in the biomedical sciences and chemistry.
Chosen by an advisory board of eminent scientists, Searle Scholars are considered among the most creative researchers pursuing careers in academic research. Their investigations address challenging research questions and can lead to new insights that fundamentally change their fields—and to opportunities for translating discoveries into new therapeutics and diagnostics.
“I am truly grateful for the support of the Searle Scholar Program as we embark on this ambitious project,” says Hrvatin, who joined the Institute in 2021 and is also an assistant professor of biology at Massachusetts Institute of Technology. The three-year grant accompanying the award will support his work developing a new animal model for the study of hibernation.
“The ability to maintain nearly constant body temperature is a defining feature of mammalian and avian evolution; but, when challenged by harsh environments, many species decrease body temperature and metabolic rate and initiate energy-conserving states of torpor and hibernation,” Hrvatin notes. “Science has not yet answered the fundamental questions of how mammals initiate, regulate, and survive these extraordinary hypometabolic and hypothermic states.
“However, those answers could have profound medical applications,” he explains. “For example, harnessing the mechanisms behind hibernation might provide new approaches to protect neurons from ischemic injury and to preserve tissues and organs for transplantation.”
In the Searle-supported study, Hrvatin aims to discover a control center in the brain that regulates distinct stages of hibernation in the Syrian hamster. His lab will start by identifying the brain regions active during the deep torpor stage of hibernation and, using molecular profiling techniques, will then identify the specific neuronal populations and molecular pathways involved. Finally, the team will develop new tools to determine specific activities in those neural populations that are necessary for natural hibernation—and that may be sufficient to induce a synthetic state of hibernation.
“Taken together,” Hrvatin says, “I believe that our discoveries and the tools we build will help establish the first controllable animal model of hibernation.”
Since 1981, 677 scientists have been named Searle Scholars and the Program has awarded more than $152 million in support for Scholars’ research. To date, 85 Searle Scholars have been inducted into the National Academy of Sciences, 20 have been recognized with a MacArthur Fellowship, and two have been awarded the Nobel Prize for Chemistry.
Genomic studies of cancer patients have revealed thousands of mutations linked to tumor development. However, for the vast majority of those mutations, researchers are unsure of how they contribute to cancer because there’s no easy way to study them in animal models.
In an advance that could help scientists make a dent in that long list of unexplored mutations, MIT researchers have developed a way to easily engineer specific cancer-linked mutations into mouse models.
Using this technique, which is based on CRISPR genome-editing technology, the researchers have created models of several different mutations of the cancer-causing gene Kras, in different organs. They believe this technique could also be used for nearly any other type of cancer mutation that has been identified.
Such models could help researchers identify and test new drugs that target these mutations.
“This is a remarkably powerful tool for examining the effects of essentially any mutation of interest in an intact animal, and in a fraction of the time required for earlier methods,” says Tyler Jacks, the David H. Koch Professor of Biology, a member of the Koch Institute for Integrative Cancer Research at MIT, and one of the senior authors of the new study.
Francisco Sánchez-Rivera, an assistant professor of biology at MIT and member of the Koch Institute, and David Liu, a professor in the Harvard University Department of Chemistry and Chemical Biology and a core institute member of the Broad Institute, are also senior authors of the study, which appears today in Nature Biotechnology.
Zack Ely PhD ’22, a former MIT graduate student who is now a visiting scientist at MIT, and MIT graduate student Nicolas Mathey-Andrews are the lead authors of the paper.
Faster editing
Testing cancer drugs in mouse models is an important step in determining whether they are safe and effective enough to go into human clinical trials. Over the past 20 years, researchers have used genetic engineering to create mouse models by deleting tumor suppressor genes or activating cancer-promoting genes. However, this approach is labor-intensive and requires several months or even years to produce and analyze mice with a single cancer-linked mutation.
“A graduate student can build a whole PhD around building a model for one mutation,” Ely says. “With traditional models, it would take the field decades to catch up to all of the mutations we’ve discovered with the Cancer Genome Atlas.”
In the mid-2010s, researchers began exploring the possibility of using the CRISPR genome-editing system to make cancerous mutations more easily. Some of this work occurred in Jacks’ lab, where Sánchez-Rivera (then an MIT graduate student) and his colleagues showed that they could use CRISPR to quickly and easily knock out genes that are often lost in tumors. However, while this approach makes it easy to knock out genes, it doesn’t lend itself to inserting new mutations into a gene because it relies on the cell’s DNA repair mechanisms, which tend to introduce errors.
Inspired by research from Liu’s lab at the Broad Institute, the MIT team wanted to come up with a way to perform more precise gene-editing that would allow them to make very targeted mutations to either oncogenes (genes that drive cancer) or tumor suppressors.
In 2019, Liu and colleagues reported a new version of CRISPR genome-editing called prime editing. Unlike the original version of CRISPR, which uses an enzyme called Cas9 to create double-stranded breaks in DNA, prime editing uses a modified enzyme called Cas9 nickase, which is fused to another enzyme called reverse transcriptase. This fusion enzyme cuts only one strand of the DNA helix, which avoids introducing double-stranded DNA breaks that can lead to errors when the cell repairs the DNA.
The MIT researchers designed their new mouse models by engineering the gene for the prime editor enzyme into the germline cells of the mice, which means that it will be present in every cell of the organism. The encoded prime editor enzyme allows cells to copy an RNA sequence into DNA that is incorporated into the genome. However, the prime editor gene remains silent until activated by the delivery of a specific protein called Cre recombinase.
Since the prime editing system is installed in the mouse genome, researchers can initiate tumor growth by injecting Cre recombinase into the tissue where they want a cancer mutation to be expressed, along with a guide RNA that directs Cas9 nickase to make a specific edit in the cells’ genome. The RNA guide can be designed to induce single DNA base substitutions, deletions, or additions in a specified gene, allowing the researchers to create any cancer mutation they wish.
Modeling mutations
To demonstrate the potential of this technique, the researchers engineered several different mutations into the Kras gene, which drives about 30 percent of all human cancers, including nearly all pancreatic adenocarcinomas. However, not all Kras mutations are identical. Many Kras mutations occur at a location known as G12, where the amino acid glycine is found, and depending on the mutation, this glycine can be converted into one of several different amino acids.
The researchers developed models of four different types of Kras mutations found in lung cancer: G12C, G12D, G12R, and G12A. To their surprise, they found that the tumors generated in each of these models had very different traits. For example, G12R mutations produced large, aggressive lung tumors, while G12A tumors were smaller and progressed more slowly.
Learning more about how these mutations affect tumor development differently could help researchers develop drugs that target each of the different mutations. Currently, there are only two FDA-approved drugs that target Kras mutations, and they are both specific to the G12C mutation, which accounts for about 30 percent of the Kras mutations seen in lung cancer.
The researchers also used their technique to create pancreatic organoids with several different types of mutations in the tumor suppressor gene p53, and they are now developing mouse models of these mutations. They are also working on generating models of additional Kras mutations, along with other mutations that help to confer resistance to Kras inhibitors.
“One thing that we’re excited about is looking at combinations of mutations including Kras mutations that drives tumorigenesis, along with resistance associated mutations,” Mathey-Andrews says. “We hope that will give us a handle on not just whether the mutation causes resistance, but what does a resistant tumor look like?”
The researchers have made mice with the prime editing system engineered into their genome available through a repository at the Jackson Laboratory, and they hope that other labs will begin to use this technique for their own studies of cancer mutations.
The research was funded by the Ludwig Center at MIT, the National Cancer Institute, a Howard Hughes Medical Institute Hanna Grey Fellowship, the V Foundation for Cancer Research, a Koch Institute Frontier Award, the MIT Research Support Committee, a Helen Hay Whitney Postdoctoral Fellowship, the David H. Koch Graduate Fellowship Fund, the National Institutes of Health, and the Lustgarten Foundation for Pancreatic Cancer Research.
Other authors of the paper include Santiago Naranjo, Samuel Gould, Kim Mercer, Gregory Newby, Christina Cabana, William Rideout, Grissel Cervantes Jaramillo, Jennifer Khirallah, Katie Holland, Peyton Randolph, William Freed-Pastor, Jessie Davis, Zachary Kulstad, Peter Westcott, Lin Lin, Andrew Anzalone, Brendan Horton, Nimisha Pattada, Sean-Luc Shanahan, Zhongfeng Ye, Stefani Spranger, and Qiaobing Xu.

According to MIT’s charter, established in 1861, part of the Institute’s mission is to advance the “development and practical application of science in connection with arts, agriculture, manufactures, and commerce.” Today, the Abdul Latif Jameel Water and Food Systems Lab (J-WAFS) is one of the driving forces behind water and food-related research on campus, much of which relates to agriculture. In 2022, J-WAFS established the Water and Food Grand Challenge Grant to inspire MIT researchers to work toward a water-secure and food-secure future for our changing planet. Not unlike MIT’s Climate Grand Challenges, the J-WAFS Grand Challenge seeks to leverage multiple areas of expertise, programs, and Institute resources. The initial call for statements of interests returned 23 letters from MIT researchers spanning 18 departments, labs, and centers. J-WAFS hosted workshops for the proposers to present and discuss their initial ideas. These were winnowed down to a smaller set of invited concept papers, followed by the final proposal stage.
Today, J-WAFS is delighted to report that the inaugural J-WAFS Grand Challenge Grant has been awarded to a team of researchers led by Professor Matt Shoulders and research scientist Robert Wilson of the Department of Chemistry. A panel of expert, external reviewers highly endorsed their proposal, which tackles a longstanding problem in crop biology — how to make photosynthesis more efficient. The team will receive $1.5 million over three years to facilitate a multistage research project that combines cutting-edge innovations in synthetic and computational biology. If successful, this project could create major benefits for agriculture and food systems worldwide.
“Food systems are a major source of global greenhouse gas emissions, and they are also increasingly vulnerable to the impacts of climate change. That’s why when we talk about climate change, we have to talk about food systems, and vice versa,” says Maria T. Zuber, MIT’s vice president for research. “J-WAFS is central to MIT’s efforts to address the interlocking challenges of climate, water, and food. This new grant program aims to catalyze innovative projects that will have real and meaningful impacts on water and food. I congratulate Professor Shoulders and the rest of the research team on being the inaugural recipients of this grant.”
Shoulders will work with Bryan Bryson, associate professor of biological engineering, as well as Bin Zhang, associate professor of chemistry, and Mary Gehring, a professor in the Department of Biology and the Whitehead Institute for Biomedical Research. Robert Wilson from the Shoulders lab will be coordinating the research effort. The team at MIT will work with outside collaborators Spencer Whitney, a professor from the Australian National University, and Ahmed Badran, an assistant professor at the Scripps Research Institute. A milestone-based collaboration will also take place with Stephen Long, a professor from the University of Illinois at Urbana-Champaign. The group consists of experts in continuous directed evolution, machine learning, molecular dynamics simulations, translational plant biochemistry, and field trials.
“This project seeks to fundamentally improve the RuBisCO enzyme that plants use to convert carbon dioxide into the energy-rich molecules that constitute our food,” says J-WAFS Director John H. Lienhard V. “This difficult problem is a true grand challenge, calling for extensive resources. With J-WAFS’ support, this long-sought goal may finally be achieved through MIT’s leading-edge research,” he adds.
RuBisCO: No, it’s not a new breakfast cereal; it just might be the key to an agricultural revolution
A growing global population, the effects of climate change, and social and political conflicts like the war in Ukraine are all threatening food supplies, particularly grain crops. Current projections estimate that crop production must increase by at least 50 percent over the next 30 years to meet food demands. One key barrier to increased crop yields is a photosynthetic enzyme called Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase (RuBisCO). During photosynthesis, crops use energy gathered from light to draw carbon dioxide (CO2) from the atmosphere and transform it into sugars and cellulose for growth, a process known as carbon fixation. RuBisCO is essential for capturing the CO2 from the air to initiate conversion of CO2 into energy-rich molecules like glucose. This reaction occurs during the second stage of photosynthesis, also known as the Calvin cycle. Without RuBisCO, the chemical reactions that account for virtually all carbon acquisition in life could not occur.
Unfortunately, RuBisCO has biochemical shortcomings. Notably, the enzyme acts slowly. Many other enzymes can process a thousand molecules per second, but RuBisCO in chloroplasts fixes less than six carbon dioxide molecules per second, often limiting the rate of plant photosynthesis. Another problem is that oxygen (O2) molecules and carbon dioxide molecules are relatively similar in shape and chemical properties, and RuBisCO is unable to fully discriminate between the two. The inadvertent fixation of oxygen by RuBisCO leads to energy and carbon loss. What’s more, at higher temperatures RuBisCO reacts even more frequently with oxygen, which will contribute to decreased photosynthetic efficiency in many staple crops as our climate warms.
The scientific consensus is that genetic engineering and synthetic biology approaches could revolutionize photosynthesis and offer protection against crop losses. To date, crop RuBisCO engineering has been impaired by technological obstacles that have limited any success in significantly enhancing crop production. Excitingly, genetic engineering and synthetic biology tools are now at a point where they can be applied and tested with the aim of creating crops with new or improved biological pathways for producing more food for the growing population.
An epic plan for fighting food insecurity
The 2023 J-WAFS Grand Challenge project will use state-of-the-art, transformative protein engineering techniques drawn from biomedicine to improve the biochemistry of photosynthesis, specifically focusing on RuBisCO. Shoulders and his team are planning to build what they call the Enhanced Photosynthesis in Crops (EPiC) platform. The project will evolve and design better crop RuBisCO in the laboratory, followed by validation of the improved enzymes in plants, ultimately resulting in the deployment of enhanced RuBisCO in field trials to evaluate the impact on crop yield.
Several recent developments make high-throughput engineering of crop RuBisCO possible. RuBisCO requires a complex chaperone network for proper assembly and function in plants. Chaperones are like helpers that guide proteins during their maturation process, shielding them from aggregation while coordinating their correct assembly. Wilson and his collaborators previously unlocked the ability to recombinantly produce plant RuBisCO outside of plant chloroplasts by reconstructing this chaperone network in Escherichia coli (E. coli). Whitney has now established that the RuBisCO enzymes from a range of agriculturally relevant crops, including potato, carrot, strawberry, and tobacco, can also be expressed using this technology. Whitney and Wilson have further developed a range of RuBisCO-dependent E. coli screens that can identify improved RuBisCO from complex gene libraries. Moreover, Shoulders and his lab have developed sophisticated in vivo mutagenesis technologies that enable efficient continuous directed evolution campaigns. Continuous directed evolution refers to a protein engineering process that can accelerate the steps of natural evolution simultaneously in an uninterrupted cycle in the lab, allowing for rapid testing of protein sequences. While Shoulders and Badran both have prior experience with cutting-edge directed evolution platforms, this will be the first time directed evolution is applied to RuBisCO from plants.
Artificial intelligence is changing the way enzyme engineering is undertaken by researchers. Principal investigators Zhang and Bryson will leverage modern computational methods to simulate the dynamics of RuBisCO structure and explore its evolutionary landscape. Specifically, Zhang will use molecular dynamics simulations to simulate and monitor the conformational dynamics of the atoms in a protein and its programmed environment over time. This approach will help the team evaluate the effect of mutations and new chemical functionalities on the properties of RuBisCO. Bryson will employ artificial intelligence and machine learning to search the RuBisCO activity landscape for optimal sequences. The computational and biological arms of the EPiC platform will work together to both validate and inform each other’s approaches to accelerate the overall engineering effort.
Shoulders and the group will deploy their designed enzymes in tobacco plants to evaluate their effects on growth and yield relative to natural RuBisCO. Gehring, a plant biologist, will assist with screening improved RuBisCO variants using the tobacco variety Nicotiana benthamianaI, where transient expression can be deployed. Transient expression is a speedy approach to test whether novel engineered RuBisCO variants can be correctly synthesized in leaf chloroplasts. Variants that pass this quality-control checkpoint at MIT will be passed to the Whitney Lab at the Australian National University for stable transformation into Nicotiana tabacum (tobacco), enabling robust measurements of photosynthetic improvement. In a final step, Professor Long at the University of Illinois at Urbana-Champaign will perform field trials of the most promising variants.
Even small improvements could have a big impact
A common criticism of efforts to improve RuBisCO is that natural evolution has not already identified a better enzyme, possibly implying that none will be found. Traditional views have speculated a catalytic trade-off between RuBisCO’s specificity factor for CO2 / O2 versus its CO2 fixation efficiency, leading to the belief that specificity factor improvements might be offset by even slower carbon fixation or vice versa. This trade-off has been suggested to explain why natural evolution has been slow to achieve a better RuBisCO. But Shoulders and the team are convinced that the EPiC platform can unlock significant overall improvements to plant RuBisCO. This view is supported by the fact that Wilson and Whitney have previously used directed evolution to improve CO2 fixation efficiency by 50 percent in RuBisCO from cyanobacteria (the ancient progenitors of plant chloroplasts) while simultaneously increasing the specificity factor.
The EPiC researchers anticipate that their initial variants could yield 20 percent increases in RuBisCO’s specificity factor without impairing other aspects of catalysis. More sophisticated variants could lift RuBisCO out of its evolutionary trap and display attributes not currently observed in nature. “If we achieve anywhere close to such an improvement and it translates to crops, the results could help transform agriculture,” Shoulders says. “If our accomplishments are more modest, it will still recruit massive new investments to this essential field.”
Successful engineering of RuBisCO would be a scientific feat of its own and ignite renewed enthusiasm for improving plant CO2 fixation. Combined with other advances in photosynthetic engineering, such as improved light usage, a new green revolution in agriculture could be achieved. Long-term impacts of the technology’s success will be measured in improvements to crop yield and grain availability, as well as resilience against yield losses under higher field temperatures. Moreover, improved land productivity together with policy initiatives would assist in reducing the environmental footprint of agriculture. With more “crop per drop,” reductions in water consumption from agriculture would be a major boost to sustainable farming practices.
“Our collaborative team of biochemists and synthetic biologists, computational biologists, and chemists is deeply integrated with plant biologists and field trial experts, yielding a robust feedback loop for enzyme engineering,” Shoulders adds. “Together, this team will be able to make a concerted effort using the most modern, state-of-the-art techniques to engineer crop RuBisCO with an eye to helping make meaningful gains in securing a stable crop supply, hopefully with accompanying improvements in both food and water security.”

Two faculty members from the MIT Department of Biology have been selected by the Howard Hughes Medical Institute (HHMI) for the inaugural cohort of HHMI Freeman Hrabowski Scholars.
Seychelle Vos, the Robert A. Swanson Career Development Professor of Life Sciences, and Hernandez Moura Silva, an assistant professor of biology and core member of the Ragon Institute of MGH, MIT and Harvard, are among 31 early-career faculty selected for their potential to become leaders in their research fields and to create diverse and inclusive lab environments in which everyone can thrive, according to a press release.
Freeman Hrabowski Scholars are appointed to a five-year term, renewable for a second five-year term after a successful progress evaluation. Each scholar will receive up to $8.6 million over 10 years, including full salary, benefits, a research budget, and scientific equipment. In addition, they will participate in professional development to advance their leadership and mentorship skills.
The Freeman Hrabowski Scholars Program represents a key component of HHMI’s diversity, equity, and inclusion goals. Over the next 20 years, HHMI expects to hire and support up to 150 Freeman Hrabowski Scholars — appointing roughly 30 scholars every other year for the next 10 years. The institute has committed up to $1.5 billion for the Freeman Hrabowski Scholars to be selected over the next decade. The program was named for Freeman A. Hrabowski III, president emeritus of the University of Maryland at Baltimore County, who played a major role in increasing the number of scientists, engineers, and physicians from backgrounds underrepresented in science in the United States.
Seychelle Vos
Seychelle Vos studies how DNA organization impacts gene expression at the atomic level, using cryogenic electron microscopy (cryo-EM), X-ray crystallography, biochemistry, and genetics. Human cells contain about 2 meters of DNA, which is packed so tightly that its entirety is contained within the nucleus, which is only a few microns across. Although DNA needs to be compacted, it also needs to be accessible to, and readable by, the cell’s molecular machinery.
Vos received a BS in genetics from the University of Georgia in 2008 and a PhD from University of California at Berkeley in 2013. During her postdoctoral research at the Max Planck Institute for Biophysical Chemistry in Germany, she determined how the molecular machine responsible for gene expression is regulated near gene promoters.
Vos joined MIT as an assistant professor of biology in fall 2019.
“I am very humbled and honored to have been named a HHMI Freeman Hrabowski Scholar,” Vos says. “It would not have been possible without the hard work of my lab and the help of my colleagues. It provides us with the support to achieve our ambitious research goals.”
Hernandez Moura Silva
Hernandez Moura Silva studies the role of immune cells in the maintenance and normal function of our bodies and tissues, beyond their role in battling infection. Specifically, he looks at a specific type of immune cell called a macrophage and its role in the proper function of white adipose tissue — our fat. White adipose tissue in a healthy state is highly populated by macrophages, including very abundant ones known as “vasculature-associated adipose tissue macrophages,” which are located around the blood vessels. When the activity of these adipose macrophages is disrupted, there are changes in the proper function of the white adipose tissue, which may ultimately link to disease. By understanding macrophage function in healthy tissues, Hernandez hopes to learn how to restore tissue homeostasis in disease.
Hernandez Moura Silva received a BS in biology in 2005 and an MSc in molecular biology in 2008 from the University of Brazil. He received his PhD in 2011 from the University of São Paulo Heart Institute. Silva pursued his postdoctoral work as the Bernard Levine Postdoctoral Fellow in immunology and immuno-metabolism at the New York University School of Medicine Skirball Institute of Biomolecular Medicine.
He joined MIT as an assistant professor of biology in 2022. He is also a core member of the Ragon Institute.
“For an immigrant coming from an underrepresented group, it’s a huge privilege to be granted this opportunity from HHMI that will empower me and my lab to shape the next generation of scientists and provide an environment where people can feel welcome and encouraged to do the science that they love and be successful,” Silva says. “It also aligns with MIT’s commitment to increase diversity and opportunity across the Institute and to become a place where all people can thrive.”

Eight MIT faculty members are among more than 250 leaders from academia, the arts, industry, public policy, and research elected to the American Academy of Arts and Sciences, the academy announced April 19.
One of the nation’s most prestigious honorary societies, the academy is also a leading center for independent policy research. Members contribute to academy publications, as well as studies of science and technology policy, energy and global security, social policy and American institutions, the humanities and culture, and education.
Those elected from MIT in 2023 are:
“With the election of these members, the academy is honoring excellence, innovation, and leadership and recognizing a broad array of stellar accomplishments. We hope every new member celebrates this achievement and joins our work advancing the common good,” says David W. Oxtoby, president of the academy.
Since its founding in 1780, the academy has elected leading thinkers from each generation, including George Washington and Benjamin Franklin in the 18th century, Maria Mitchell and Daniel Webster in the 19th century, and Toni Morrison and Albert Einstein in the 20th century. The current membership includes more than 250 Nobel and Pulitzer Prize winners.

On April 14 2003, scientists announced the end to one of the most remarkable achievements in history: the first (nearly) complete sequencing of a human genome. It was the culmination of a decade-plus endeavor that involved thousands of scientists across the globe. Many people hoped the accomplishment would change the world for the better.
For the 20-year anniversary of this historic event, we took a look back at the Human Genome Project and its impact. How did it shape science moving forward? How many of the expected goals have been reached since? And what lies ahead for the study of genetics?
A genome is the entire set of genetic information that makes up an organism. This information is packaged into sequences of DNA we call genes, which in humans are spread along 23 pairs of chromosomes. Only a small portion of these genes contain the instructions for coding the many proteins essential for life, but much of the rest is still thought to be important to our functioning. As scientists would eventually confirm, one copy of the human genome has around 3 billion base pairs of DNA. The sheer magnitude of the effort needed to map all this wasn’t lost on the researchers involved in the project, especially given the technology available decades ago.
“There’s been lots of analogies that people have put forward—like us being Lewis and Clark. We didn’t really have a map,” said Richard Gibbs, founder and director of the Baylor College of Medicine Human Genome Sequencing Center in Texas, one of the major institutions involved in the project.
Gibbs and his many colleagues knew they had to make compromises. Despite the advancements in sequencing technology since the official start of the project in 1990, they couldn’t fill in every gap with their current tools. And the first human genome was a composite of several blood donors in the U.S., not a single person. Along the way, private company Celera entered the picture, promising that it would complete a separate genome project using its own techniques even faster. Ultimately, both groups finished ahead of schedule around the same time, with the first draft sequences released in 2000, though Celera announced its success a few months earlier.
Regardless of the victor, the feat certainly did usher in a new era of genetics research—one that has seen great leaps in speed and efficiency since 2003.
“I think the very most important accomplishment in the past 20 years has been the advent of next-generation sequencing. The ability to perform sequencing in a massively parallel way, so that you could do it far more quickly and cheaply,” said Stacey Gabriel, director of the Genomics Platform at the Broad Institute of MIT and Harvard, another major research site involved in the Human Genome Project. “And that has come with all of the associated advancement in our computational abilities, too, to really be able to take that data and analyze it at a massive scale as well.”
The original project cost $2.7 billion, with most of the genome being mapped over a two-year span. Nowadays, the current speed record for sequencing a genome is around five hours (more often, though, it takes weeks), and this past fall, the company Illumina unveiled a machine that it claims will cost as little as $200 per sequence, down from the recently typical $600 cost.
Greg Findlay leads the Genome Function Laboratory at the Francis Crick Institute in the UK. His team is one of many around the world that is building on the work of the original project. They’re currently trying to identify and understand how certain variants in tumor-suppressor genes can raise our risk of cancer.
“So what my lab tries to do is actually understand what variants do to the genome. That is, we want to know exactly which variants cause disease and why they cause disease. Right now, we’re focused on certain genetic changes that lead to cancer, and I think this particular field has really been revolutionized by the Human Genome Project,” Findlay said. “We now know there are many, many different genetic paths by which cells can turn into cancer. And we know this, because we’ve been able to actually sequence the DNA in so many different tumors repeatedly, across all different types of cancer, to see what are the mutations that actually lead to cancer forming.”
Perhaps equally important was the project’s impact on scientific collaboration. The effort directly led to an international agreement meant to ensure open access to DNA sequences. It also made clear that great things could be possible when large groups of scientists worked together, according to Gibbs.
“It simply changed the way that people thought that biology could be done,” he said. “It built a model for team science that was not there before.”
Even at the time, though, the project knowingly left some things unfinished. They had mapped roughly 92% of the genome by 2003, but it would take almost 20 more years for other scientists to track down the remaining 8%. This missing “dark matter” of our genome could very well provide new clues about how humans evolved or our susceptibility to various diseases.
Much of the genetic information collected and analyzed since the project ended has come from white and European populations—a disparity that hampers our ability to truly understand the impact of genetics on everyone’s health. But scientists today are working on bridging that gap through initiatives like the Human Pangenome Project, which will sequence and make available the full genomes of over 300 people intended to represent the breadth of human diversity around the globe.
“There’s genetic variation that exists across all the world’s populations. And if you only use variations from a sliver of the world’s populations, and you try to apply that to everybody else, it just doesn’t work very well, because we all have different backgrounds,” said Lucinda Antonacci-Fulton, one of the project’s coordinators and director of project development & new initiatives at Washington University in St. Louis’s McDonnell Genome Institute. “So the more inclusive you can be, the better off you are in terms of treatments that you want to bring into the clinic.”
As important as genetics research has been, some of the expectations fueled by the Human Genome Project likely were too lofty. In 2000, for instance, President Bill Clinton claimed that the project would “revolutionize the diagnosis, prevention and treatment of most, if not all, human diseases.” While we do continue to discover new gene variants that strongly predict the odds of developing a specific disease or trait, there are countless others that we’re still in the dark about. Elsewhere, it’s become clear that our genome often only plays a small or negligible role in why we get sick or experience something in a particular manner. So although the project has helped unlock some of the mysteries of the world, there are so many more questions out there about why we are the way we are, and our genes are probably not going to provide a neat answer to many of them.
“I think where things are oversold, sometimes, is with this notion that just because the human genome is done, you’re going to be able to read it off for some sort of deterministic answer—where finding genes for disease becomes like falling off a log,” Gabriel notes. “But often human disease, especially the diseases that impact us the most, these are not simple genetic diseases. They’re multifactorial. They’re combinations of your genes, your behavior, exposure to the environment, sometimes just bad luck.”
None of this is to sell short the potential of genetics. Hans Lehrach, a former director at the Max Planck Institute for Molecular Genetics in Germany, was one of the first researchers involved in the Human Genome Project. He’s also one of many scientists who believe that we’ll someday be able to cheaply and easily scan a person’s genome at a moment’s notice and that this information, along with other aspects of our molecular make-up, will help guide the specific drugs or interventions doctors prescribe—a concept known as personalized medicine. Notably, the treatment of some cancers is already influenced by the variants that underlie their growth. Some experts even argue that widespread whole genome sequencing should start as early as birth, and there are already small-scale programs in the U.S., UK, and elsewhere testing out its potential benefits and risks.
“Not knowing about your genome is a bit like crossing the street while closing your eyes because you don’t want to see a bus coming. If we don’t sequence our genomes, the buses keep coming anyway—it just lets us open our eyes and maybe see the kind of danger that we can escape or do something about,” Lehrach said.
The Human Genome Project truly has changed the scientific landscape, but we’re still only at the very beginning of seeing the world that it’s made possible.