Rest in pieces: deconstructing polypeptide degradation machinery
Lillian Eden | Department of Biology
November 12, 2024
Research from the Sauer and Davis Labs in the Department of Biology at MIT shows that conformational changes contribute to the specificity of “molecular woodchippers”
Degradation is a crucial process for maintaining protein homeostasis by culling excess or damaged proteins whose components can then be recycled. It is also a highly regulated process—for good reason. A cell could potentially waste many resources if the degradation machinery destroys proteins it shouldn’t.
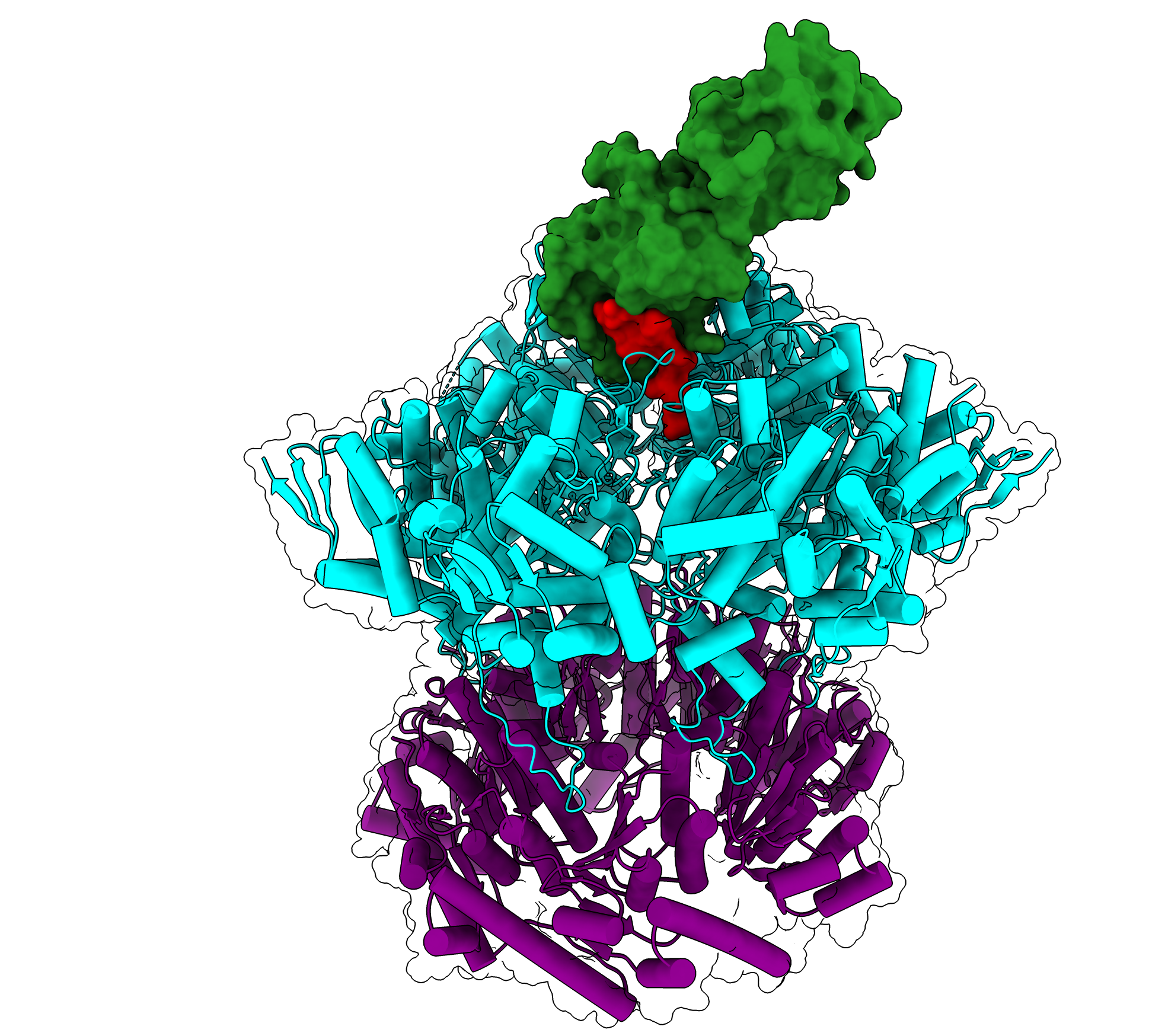
One of the major pathways for protein degradation in bacteria and eukaryotic mitochondria involves a molecular machine called ClpXP. ClpXP is made up of two components: a star-shaped structure made up of six subunits called ClpX that engages and unfolds proteins tagged for degradation, and an associated barrel-shaped enzyme, called ClpP, that chemically breaks up proteins into small pieces called peptides.
ClpXP is incredibly adaptable and is often compared to a woodchipper — able to take in materials and spit out their broken-down components. Thanks to biochemical experiments, this molecular degradation machine is known to be able to break down hundreds of different proteins in the cell regardless of physical or chemical properties such as size, shape, or charge. ClpX uses energy from ATP hydrolysis to unfold proteins before they are threaded through its central channel, referred to as the axial channel, and into the degradation chamber of ClpP.
In three papers, one in PNAS and two in Nature Communications, researchers from the Department of Biology at MIT have expanded our understanding of how this molecular machinery engages with, unfolds, and degrades proteins — and how that machinery refrains, by design, from unfolding proteins not tagged for degradation.
Alireza Ghanbarpour, until recently a postdoc in the Sauer Lab and Davis Lab and first author on all three papers, began with a simple question: given the vast repertoire of potential substrates — that is, proteins to be degraded — how is ClpXP so specific?
Ghanbarpour — now an assistant professor in the Department of Biochemistry and Molecular Biology at Washington University School of Medicine in St. Louis — found that the answer to this question lies in conformational changes in the molecular machine as it engages with an ill-fated protein.
Reverse Engineering using Structural Insights
Ghanbarpour approached the question of ClpXP’s versatility by characterizing conformational changes of the molecular machine using a technique called cryogenic electron microscopy. In cryo-EM, sample particles are frozen in solution, and images are collected; algorithms then create 3D renderings from the 2D images.
“It’s really useful to generate different structures in different conditions and then put them together until you know how a machine works,” he says. “I love structural biology, and these molecular machines make fascinating targets for structural work and biochemistry. Their structural plasticity and precise functions offer exciting opportunities to understand how nature leverages enzyme conformations to generate novel functions and tightly regulate protein degradation within the cell.”
Inside the cell, these proteases do not work alone but instead work together with “adaptor” proteins, which can promote — or inhibit — degradation by ClpXP. One of the adaptor proteins that promotes degradation by ClpXP is SspB.
In E. coli and most other bacteria, ClpXP and SspB interact with a tag called ssrA that is added to incomplete proteins when their biosynthesis on ribosomes stalls.
The tagging process frees up the ribosome to make more proteins, but creates a problem: incomplete proteins are prone to aggregation, which could be detrimental to cellular health and can lead to disease. By interacting with the degradation tag, ClpXP and SspB help to ensure the degradation of these incomplete proteins. Understanding this process and how it may go awry may open therapeutic avenues in the future.
“It wasn’t clear how certain adapters were interacting with the substrate and the molecular machines during substrate delivery,” Ghanbarpour notes. “My recent structure reveals that the adapter engages with the enzyme, reaching deep into the axial channel to deliver the substrate.”
Ghanbarpour and colleagues showed that ClpX engages with both the SspB adaptor and the ssrA degradation tag of an ill-fated protein at the same time. Surprisingly, they also found that this interaction occurs while the upper part of the axial channel through ClpX is closed — in fact, the closed channel allows ClpX to contact both the tag and the adaptor simultaneously.
This result was surprising, according to senior author and Salvador E. Luria Professor of Biology Robert Sauer, whose lab has been working on understanding this molecular machine for more than two decades: it was unclear whether the channel through ClpX closes in response to a substrate interaction, or if the channel is always closed until it opens to pass an unfolded protein down to ClpP to be degraded.
Preventing Rogue Degradation
Throughout this project, Ghanbarpour was co-advised by structural biologist and Associate Professor of Biology Joey Davis and collaborated with members of the Davis Lab to better understand the conformational changes that allow these molecular machines to function. Using a cryo-EM analysis approach developed in the Davis lab called CryoDRGN, the researchers showed that there is an equilibrium between ClpXP in the open and closed states: it’s usually closed but is open in about 10% of the particles in their samples.
The closed state is almost identical to the conformation ClpXP assumes when it is engaged with an ssrA-tagged substrate and the SspB adaptor.
To better understand the biological significance of this equilibrium, Ghanbarpour created a mutant of ClpXP that is always in the open position. Compared to normal ClpXP, the mutant degraded some proteins lacking obvious degradation tags faster but degraded ssrA-tagged proteins more slowly.
According to Ghanbarpour, these results indicate that the closed channel improves ClpXP’s ability to efficiently engage tagged proteins meant to be degraded, whereas the open channel allows more “promiscuous” degradation.
Pausing the Process
The next question Ghanbarpour wanted to answer was what this molecular machine looks like while engaged with a protein it is attempting to unfold. To do that, he created a substrate with a highly stable protein attached to the degradation tag that is initially pulled into ClpX, but then dramatically slows protein unfolding and degradation.
In the structures where the degradation process stalls, Ghanbarpour found that the degradation tag was pulled far into the molecular machine—through ClpX and into ClpP—and the folded protein part of the substrate was pulled tightly against the axial channel of ClpX.
The opening of the axial channel, called the axial pore, is made up of looping protein structures called RKH loops. These flexible loops were found to play roles both in recognizing the ssrA degradation tag and in how substrates or the SspB adaptor interact with or are pulled against the channel during degradation.
The flexibility of these RKH loops allows ClpX to interact with a large number of different proteins and adapters, and these results clarify some previous biochemical and mutational studies of interactions between the substrate and ClpXP.
Although Ghanbarpour’s recent work focused on just one adaptor and degradation tag, he noted there are many more targets — ClpXP is something akin to a Swiss army knife for breaking down polypeptide chains.
The way those other substrates interact with ClpXP could differ from the structures solved with the SspB adaptor and ssrA tag. It also stands to reason that the way ClpXP reacts to each substrate may be unique. For example, given that ClpX is occasionally in an open state, some substrates may engage with ClpXP only while it’s in an open conformation.
In his new position at Washington University, Ghanbarpour intends to continue exploring how ClpXP and other molecular machines locate their target substrates and interact with adaptors, shedding light on how cells regulate protein degradation and maintain protein homeostasis.
The structures Ghanbarpour solved involved free-floating protein degradation machinery, but membrane-bound degradation machinery also exists. The membrane-bound version’s structure and conformational adaptions potentially differ from the structures Ghanbarpour found in his previous three papers. Indeed, in a recent preprint, Ghanbarpour worked on the cryo-EM structure of a nautilus shell-shaped protein assembly that seems to control membrane-bound degradation machinery. This assembly plays a critical role in regulating protein degradation within the bacterial inner membrane.
“The function of these proteases goes beyond simply degrading damaged proteins. They also target transcription factors, regulatory proteins, and proteins that don’t exist in normal conditions,” he says. “My new lab is particularly interested in understanding how cells use these proteases and their accessory adaptors, both under normal and stress conditions, to reshape the proteome and support recovery from cellular distress.”