Nicole Davis | Whitehead
November 25, 2019
A groundswell of evidence connects defects in the function of microglia, the brain’s resident immune cells, to neurodegenerative diseases, yet the tools for studying these cells in the laboratory have been limited. Now, a team of Whitehead Institute scientists has developed a new experimental platform for generating microglia from human stem cells that includes transplantation into newborn mice. As described online November 26 in the Proceedings of the National Academy of Sciences (PNAS), this new method yields microglial cells that resemble those in the human brain more closely than previous approaches, which could help enable future studies aimed at unravelling the role of microglia in neurodegeneration and other brain disorders.
“The dysfunction of microglia is implicated in a wide variety of brain conditions, and yet our knowledge of them, especially in humans, is really quite limited,” says senior author Rudolph Jaenisch, a Founding Member of the Whitehead Institute and professor of biology at the Massachusetts Institute of Technology. “This new approach will help us lift the hood on these important yet enigmatic brain cells.”
Microglia are increasingly recognized as key players in brain health and disease, but the majority of what is known about them comes from studies of mice, not humans. Yet human and mouse microglia are quite distinct — in humans, the cells are much larger, and have a more branched appearance, suggesting significant differences in their biology.
To address this gap in knowledge, multiple research teams have recently devised methods to generate microglia using human stem cells and grow them under laboratory conditions that mimic their natural environment. However, this approach has a fundamental drawback: the cultured cells do not look like microglia nor do they behave much like them, even though they display the appropriate molecular hallmarks.
“That really suggests to us that this is not the optimal approach to study how microglia are behaving in healthy and diseased brains,” says first author Devon Svoboda, a postdoctoral fellow in the Jaenisch lab. “We set out to create a new method in which the stem-cell derived microglial cells can reside in the brains of mice — one of the best models of the human brain that we have.”
Transplanting human cells into mice — creating “chimeras” — is a well-established technique. However, Svoboda and her colleagues discovered they needed to use special strains of mice that carry human genes for certain growth factors, called cytokines, which are required for microglial development and survival. The researchers utilized mice that carry human genes for four crucial cytokines: CSF1, IL3, SCF, and GM-CSF.
“What is special about these chimeras is really the mice we are using,” says Svoboda. “They express the human alleles of these cytokines which is key because the mouse versions are not able to communicate with receptors on human microglia, so the cells die.”
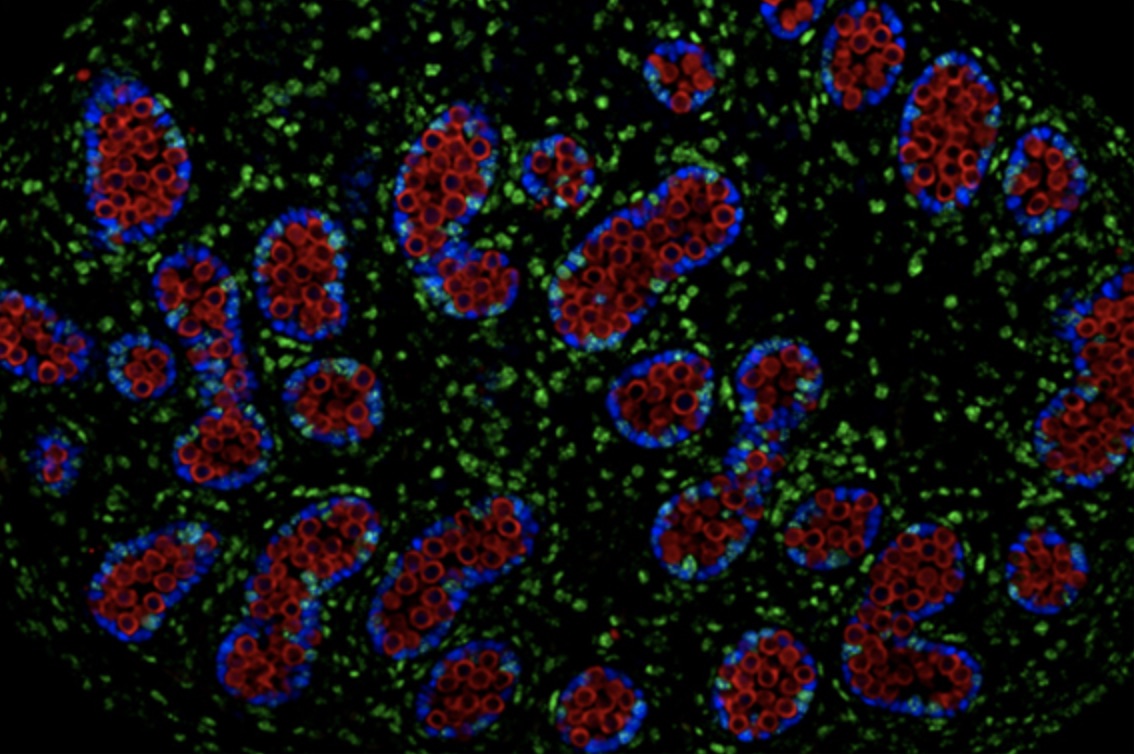
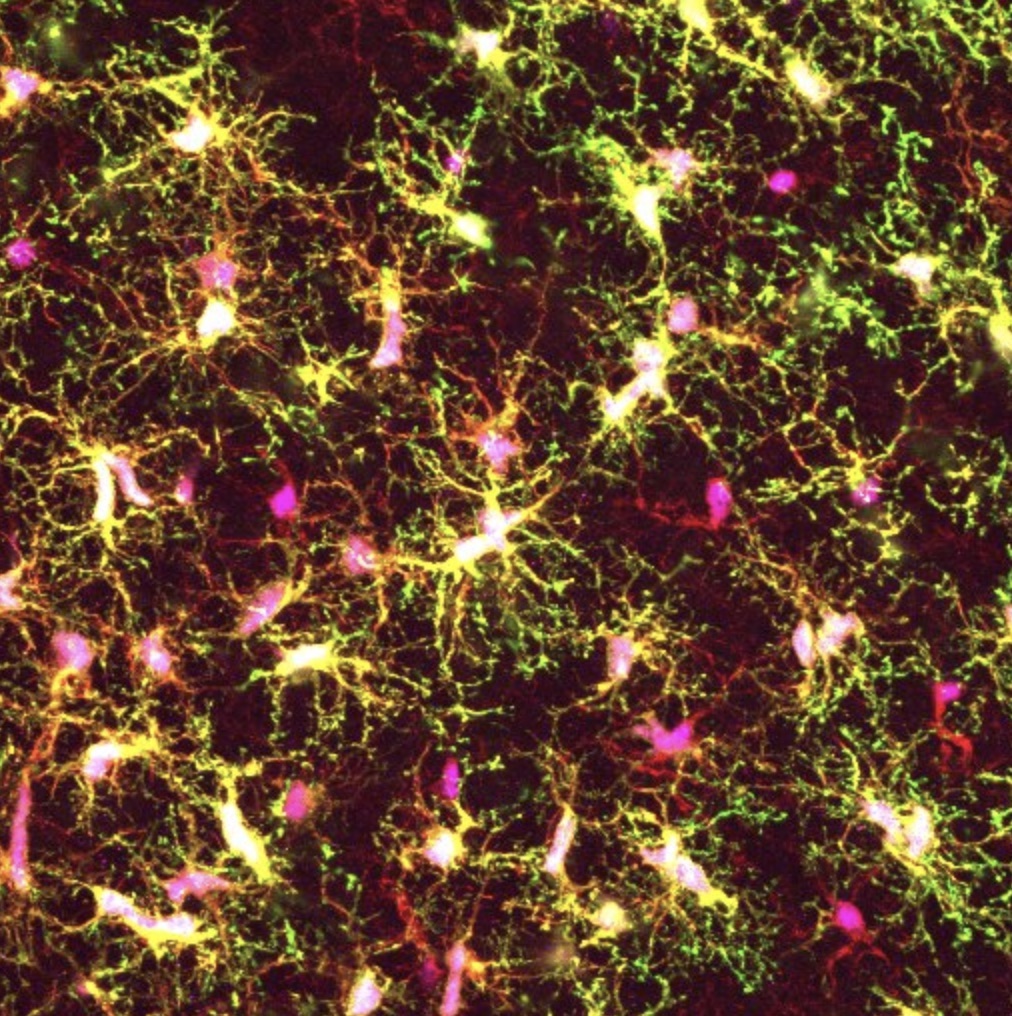
After transplanting the stem-cell derived microglia into these mice, the research team examined the cells’ morphology and their molecular characteristics. They found that the transplanted cells closely resembled those found in the human brain.
Further analyses revealed some striking differences between the team’s “chimera-grown” microglia and those grown in the laboratory using conventional cell culture methods. Surprisingly, Svoboda and her colleagues found that the cultured microglia showed strong similarities to the diseased microglia from patients with multiple sclerosis, another brain condition in which the cells are implicated.
“If you want to learn more about the role of microglia in disease, then studying them in culture is probably not the best way,” says Svoboda. “The chimeras and the in vitro methods really complement each other, and we think there is a place for both systems in microglia research going forward.”
The Whitehead-led team plans to extend their initial studies in several ways. One is to identify which cytokines and other growth factors are most crucial to microglial development. That knowledge could help improve existing cell culture methods and enable them to more closely mirror the cells’ natural environment. Another key direction is to use the new chimera-based system to create models of neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease, to understand how microglia respond to diseased neurons and, in turn, how diseased microglia can impair neuron function.
“Our chimera-based method will give us a good handle to begin to stringently test the role of microglia in brain health and disease,” says Jaenisch. “This is an important step forward for the field.”
Support for this work was provided by the Cure Alzheimer’s Foundation, MassCATS, and NIH Grants R01 AG058002-01, R01 MH104610, R37 CA084198, and U19 AI131135 (to R.J.). L.D.S. is supported by NIH Grants R24 OD26440, AI32963, and CA034196. J.S. is supported by the National Institute of Child Health and Human Development (K99HD096049).
Written by Nicole Davis
***
Rudolf Jaenisch’s primary affiliation is with Whitehead Institute for Biomedical Research, where his laboratory is located and all his research is conducted. He is also a professor of biology at Massachusetts Institute of Technology.
***
Paper cited:
1. Whitehead Institute, Cambridge, MA 02142, USA
2. Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
3.
4.