Lucy Jackub
October 17, 2019
The best available treatments for pancreatic cancer are highly toxic, and, as chemotherapies go, not very effective. The drug gemcitabine has been used for decades to extend the life of patients, but very high doses are required to combat the tumor, which grows in the pancreas surrounded by stiff, fibrous, noncancerous tissue called stroma. This hallmark of pancreatic cancer makes it unusually difficult to treat: the more stromal tissue accumulates, the less the drug works, while patients still endure brutal side effects. Only 8.5 percent of pancreatic cancer patients survive five years beyond their diagnosis, so there’s an urgent need to figure out why existing treatments are failing.
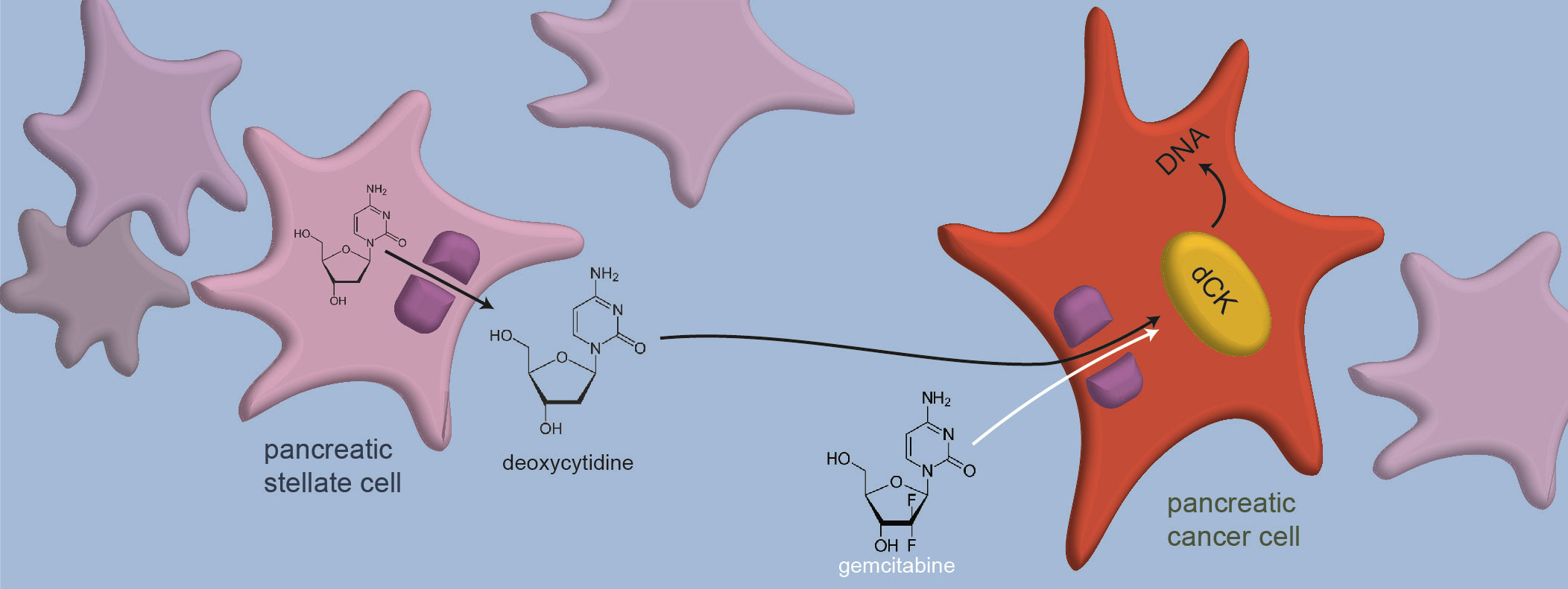
Scientists have known for a long time that gemcitabine fights cancer by killing cells during replication, though why it works for pancreatic cancer in particular is a bit of a mystery. The drug is a small molecule that masquerades as the nucleoside deoxycytidine, one unit in the nucleic acids that make up DNA. Once gemcitabine is integrated into a replicating strand of DNA, additional nucleosides can’t be joined to it. The new DNA strand can’t be completed, and the cell dies. Now, researchers from MIT have discovered that non-cancer cells in the pancreatic stromal tissue secrete astonishing quantities of deoxycytidine. They found that competition with deoxycytidine makes its imposter, gemcitabine, less effective, explaining why higher doses of the drug are needed as more stromal tissue grows around the tumor.
“That was an answer we were looking for — what is making pancreatic tumors resistant to gemcitabine?” says Michael Hemann, associate professor of biology, a member of MIT’s Koch Institute for Integrative Cancer Research, and co-senior author of the study. “Understanding the basic mechanisms of these drugs allows us to return to the clinic with improved strategies to treat patients with cancer.”
Douglas Lauffenburger, a professor of biological engineering, is also a co-senior author of the study, which represents a collaboration between the Hemann lab, the Lauffenburger lab, and the Vander Heiden lab, and appeared online in Cancer Research on September 4. Hemann lab graduate student Simona Dalin is the lead author.
The mystery ingredient
For years, researchers at MIT have been investigating different sources of chemotherapy resistance in stromal tissue. When Dalin took up the study two years ago, she was building on the findings of a former postdoc in the Hemann lab, Emanuel Kreidl. Kreidl had found that stellate cells, one type of cell in the pancreatic stromal tissue surrounding the tumor, were releasing something into the microenvironment of the pancreas that disrupted the function of gemcitabine.
Cells secrete all sorts of things — micro RNAs, fatty acids, proteins — that may be taken up and used by neighboring cells. Biologists call these ambient materials around the cell its “media.” Kreidl had tried boiling, digesting, and filtering the stellate cell media, but nothing he did made gemcitabine any more effective against the cancer cells. The usual suspects commonly implicated in drug resistance caused by neighboring cells, like proteins, would break down under such tests. “That’s when we knew there was something new here,” says Dalin. Her challenge was to figure out what that mystery ingredient was.
Mark Sullivan PhD ‘19, then a graduate student and biochemist in Vander Heiden lab, was enlisted to help separate the stellate cell media into its molecular components and identify them. After doing so, Dalin says, “it was fairly obvious that deoxycytidine was the thing that we were looking for.” Because gemcitabine works by taking deoxycytidine’s place in DNA replication, it made sense that the presence of a lot of deoxycytidine could make it difficult for gemcitabine to fulfill its function.
Molecules pass in and out of cells through gates in the cell membrane, called transporters. Using a drug that blocks certain transporters, Dalin was able to shut the gate in the stellate cells through which deoxycytidine is released. With less deoxycytidine around, the gemcitabine was effective at lower doses, confirming her hypothesis. Now, the researchers just needed to figure out how and where deoxycytidine was getting in the way of the drug.
Once inside the cell, a nucleoside must have one or more phosphate groups added to it by several enzymes in order to become a nucleotide that can be used to build DNA. Gemcitabine goes through the same process. The researchers determined that gemcitabine was competing with deoxycytidine for the first of those enzymes, deoxycytidine kinase. When they flooded the cell with that enzyme, gemcitabine didn’t have to wait in line for its phosphate groups — and could get into the DNA to work its fatal subterfuge.
Upending Assumptions
Going forward, the Hemann lab aims to identify drugs that could inhibit the production of deoxycytidine and restore the tumor’s sensitivity to gemcitabine. Senthil Muthuswamy, an associate professor of medicine at Beth Israel Deaconess Medical Center who was not involved in the research, says this study provides “new and important insights” into how and why tumors develop resistance to gemcitabine. The findings, he adds, are “likely to have important implications for developing ways to overcome gemcitabine resistance in pancreatic cancer.”
The study’s findings may shed light on other cancer treatments that work similarly to gemcitabine. For every nucleoside, there are look-alike molecules, or analogs, that are used in cancer therapies. For example, the purine analog fludarabine is used to treat acute myeloid leukemia, another tenacious carcinoma. These generic drugs have been adopted through trial and error in the clinic, but scientists don’t fully understand why they are effective at the molecular level.
In theory, nucleoside analog drugs should work interchangeably; every nucleoside is necessary in either the replication of DNA or RNA. In practice, though, these drugs are only effective for certain cancers. The MIT researchers speculate that the sheer amount of deoxycytidine being produced in the pancreas could suggest that pancreatic cells have a particular need for deoxycytidine that also makes them more responsive to its analogs — perhaps explaining why gemcitabine targets pancreatic cancer cells effectively.
“Understanding more about nucleoside biology, and more about which organs have high levels of which nucleosides, might help us understand when to use which chemotherapies,” Dalin says.
This study leaves the researchers with many questions about how and why nucleosides are produced in the body, a realm of basic biology that is still poorly understood. It’s generally assumed that cells only make nucleosides for their own internal use in DNA replication. But pancreatic stellate cells produce a lot of deoxycytidine, far more than they need for themselves, suggesting the excess nucleosides may serve some unknown purpose in neighboring cells. Although more experiments are needed to determine this mysterious purpose, the MIT researchers have some ideas.
“These extra nucleosides introduce a possibility that perhaps making deoxycytidine is a normal function of stellate cells in the pancreas, in order to provide building blocks for the cells around them,” says Hemann. “And that’s a real surprise.”
This work was funded in part by a David H. Koch Fellowship and the MIT Center for Precision Cancer Medicine.
Image: Deoxycytidine and gemcitabine, its look-alike molecule, enter a cancer cell through the same gate in the cell membrane and are altered by the same enzyme (dCK) before they are integrated into DNA. Credit: Courtesy of the researchers.
Citation:
“Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance.”
Cancer Research, online Sept. 4, 2019, DOI: 10.1158/0008-5472.CAN-19-0960.
Dalin, S., Sullivan, M.R., Lau, A.N., Grauman-Boss, B., Mueller, H.S., Kreidl, E., Fenoglio, S., Luengo, A., Lees, J.A., Vander Heiden, M.G. and Lauffenburger, D.A.