Maternal messenger RNAs (mRNAs), located within the cytoplasm of an immature egg, are crucial for jump starting development. Following fertilization, these mRNAs are passed onto the zygote, the first newly formed cell. Having been read from the maternal DNA genetic code, they serve as the sole templates for protein production essential for early development until the zygote’s own genes become active and take over.
Many maternal mRNAs are stored in ribonucleoprotein (RNP) granules, which are a type of membrane-less compartments, or condensates, within eggs and developing embryos. These granules are believed to preserve the mRNA in a “paused” state until the encoded proteins are needed for specific developmental processes upon fertilization of the egg cell. Then, certain developmental signals kick in to instruct the RNP granules to release the stored mRNA so the instructions can be translated into a functional protein.
One type of RNP granules called germ granules is found in embryo germplasm, a cytoplasmic region that gives rise to germ cells, which become the eggs or sperms of adult flies. Whitehead Institute Director Ruth Lehmann studies how germ cells form and transmit their genetic information across generations. Her lab is particularly interested in understanding how germ granules in embryos localize and regulate maternal mRNAs.
Now, Lehmann, along with graduate student Ruoyu Chen and colleagues, has uncovered that the role of germ granules in fruit flies (Drosophila melanogaster) extends beyond safeguarding maternal mRNAs. Their findings, published in the journal Nature Cell Biology on July 4, demonstrate that germ granules also play an active role in translating, or making into protein, a specific maternal mRNA, called nanos, crucial for specifying germ cells and the abdomen of the organism.
“Traditionally, scientists have thought of RNP granules as a dead zone for translation,” says Chen. “But through high-resolution imaging, we’ve challenged this notion and shown that the surface of these granules is actually a platform for translation of nanos mRNA.”
RNP granules act as vaults
Within a developing embryo, various fate-determining proteins dictate whether a cell will become a muscle, nerve, or skin cell in a fully-formed body. Nanos, a gene with conserved function in Drosophila and humans, guides the production of Nanos protein which instructs cells to develop into germline. Mutations in the nanos gene cause sterility in animals.
During early embryonic development, Nanos protein also helps establish the body plan of the fruit fly embryo — it specifies the posterior end or abdominal region, and guides the ordered development of tissues along the length of the body, from head to tail. In embryos with impaired Nanos function, the consequences are fatal.
“When Nanos protein isn’t functioning properly, the fruit fly embryos are really short,” says Chen. “This is because the embryo has no abdomen, which is basically half of its body. Nanos also has a second function that is conserved from flies to humans. This function is very local and instructs the cells with lots of Nanos to become germ cells. ”
Given Nanos’ vital role, embryos must safeguard instructions for its production until the embryo reaches a specific stage of development, when it is time to define the posterior region. Previous work has indicated that germ granules in the germplasm and germ cells can act like vaults, shielding the nanos mRNA from degradation or premature translation.
However, while the mRNA instructions for building the protein are distributed throughout the embryo, Nanos protein is found only in regions where germ granules reside. The mRNA does not get translated elsewhere in the embryo because of a regulatory protein called Smaug, named after the golden dragon depicted in J. R. R. Tolkien’s 1937 novel The Hobbit. Smaug binds to a non-protein coding segment of the mRNA known as the 3’ untranslated region (3’ UTR), extending beyond the protein-coding sequence, effectively suppressing the translation process.
For Lehmann, Chen, and their colleagues, this hinted at an intriguing relationship between nanos mRNA and germ granules. Are the granules essential for translating nanos mRNA into a functional protein? And if they are, is their role primarily to serve as a safekeeping place to evade repression by Smaug or do they actively facilitate the translation of nanos mRNA too?
To answer these questions, the researchers combined high-resolution imaging with a technique called the SunTag system to directly visualize the translation of nanos mRNA within Drosophila germ granules at the single-molecule level.
Unlike green fluorescent protein tagging, where a single fluorescent molecule is used, the SunTag system allows scientists to recruit multiple GFP copies for an amplified signal. First, a small protein tag, known as the SunTag, is fused with the protein-producing region of the nanos mRNA. As the mRNA instructions undergo translation, GFP molecules stick to the newly synthesized SunTag-Nanos protein, resulting in a bright fluorescent signal. Overlying this translation signal with fluorescent probes specifically labeling the mRNA then allows researchers to precisely visualize and track when and where the translation process is taking place.
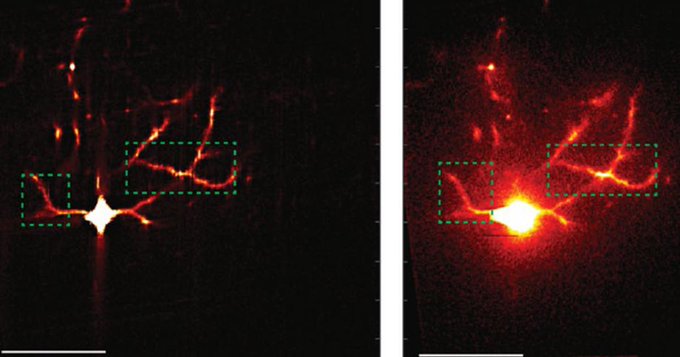
“Using this system, we’ve discovered that when nanos mRNA is translated, it protrudes slightly from the surface of the granules like snakes peeking out of a box,” says Chen. “But they can’t fully emerge; a part of their sequence, specifically their “back” end, the 3’ UTR, remains tucked inside the granules. When the RNA is not translated, like during oogenesis, the tip coils back and is hidden inside the granule.”
With their high-resolution SunTag imaging technique, Lehmann, Chen and their colleagues have directly added to the work of other researchers with similar observations: mRNAs in the process of translation are in an extended configuration, while the 5’UTR curls back to the 3’UTR when the mRNAs are repressed.
Flipping on nanos translation
Then, the researchers went on to take a closer look at how these granules help initiate translation, while Smaug is able to inhibit the same nanos mRNA molecules from being translated in other areas of the embryo. They hypothesized that the untranslated region (UTR) of nanos mRNA, which remains concealed within the granules, might be playing a pivotal role in the translation process by localizing the mRNA instructions within germ cell granules. This localization, they speculated, protects the mRNA from Smaug’s inhibitory actions and facilitates Nanos protein production, so the posterior region can develop properly.
However, counter-intuitive to a simple protection model, they found that rather than being depleted, Smaug is enriched within germ granules, indicating that additional mechanisms within the RNP granule must counteract Smaug’s inhibitory effects. To explore this, the researchers turned to another regulatory protein called Oskar, which is known to interact with Smaug.
Discovered by Lehmann in a 1986 study, and named after a character in the German novel The Tin Drum, the oskar gene in Drosophila is known to help with the development of the posterior region. Later research has revealed that, during the development of oocytes, Oskar acts as a scaffold protein by initiating the formation of germ granules in germ cells and directing mRNA molecules, including nanos, towards the granules.
To gain a deeper understanding of Oskar’s full role in translational regulation in germ granules and its interaction with Smaug, the researchers engineered a modified version of Oskar protein. This altered Oskar protein retained its ability to initiate the formation of germ granules and localize nanos mRNA within them. However, Smaug no longer localized to the germ granules assembled by this altered Oskar.
The researchers then studied whether the mutant protein had any effect on nanos mRNA translation. In the germ cells with this mutant version of Oskar, the researchers saw a significant reduction in the translation of nanos mRNA. These findings, combined, suggested that Oskar regulates nanos translation in fruit fly embryos by recruiting Smaug to the granules and then counteracting its repression of translation.
“Condensates composed of RNAs and proteins are found in the cytoplasm of pretty much every cell and are thought to mediate mRNA storage or transport,” says Lehmann, who is also a professor of biology at the Massachusetts Institute of Technology. “But our results provide new insights into condensate biology by suggesting that condensates can be also used to specifically translate stored mRNAs.”
Indeed, in the oocyte, the germ granules are silent and only become activated when the egg is fertilized.
“This suggests that there might also be other ‘on and off switches’ governing translation within condensates during early development,” adds Lehmann. “How this is achieved and whether we could engineer this to happen at will in these and other granules is a question for the future.”