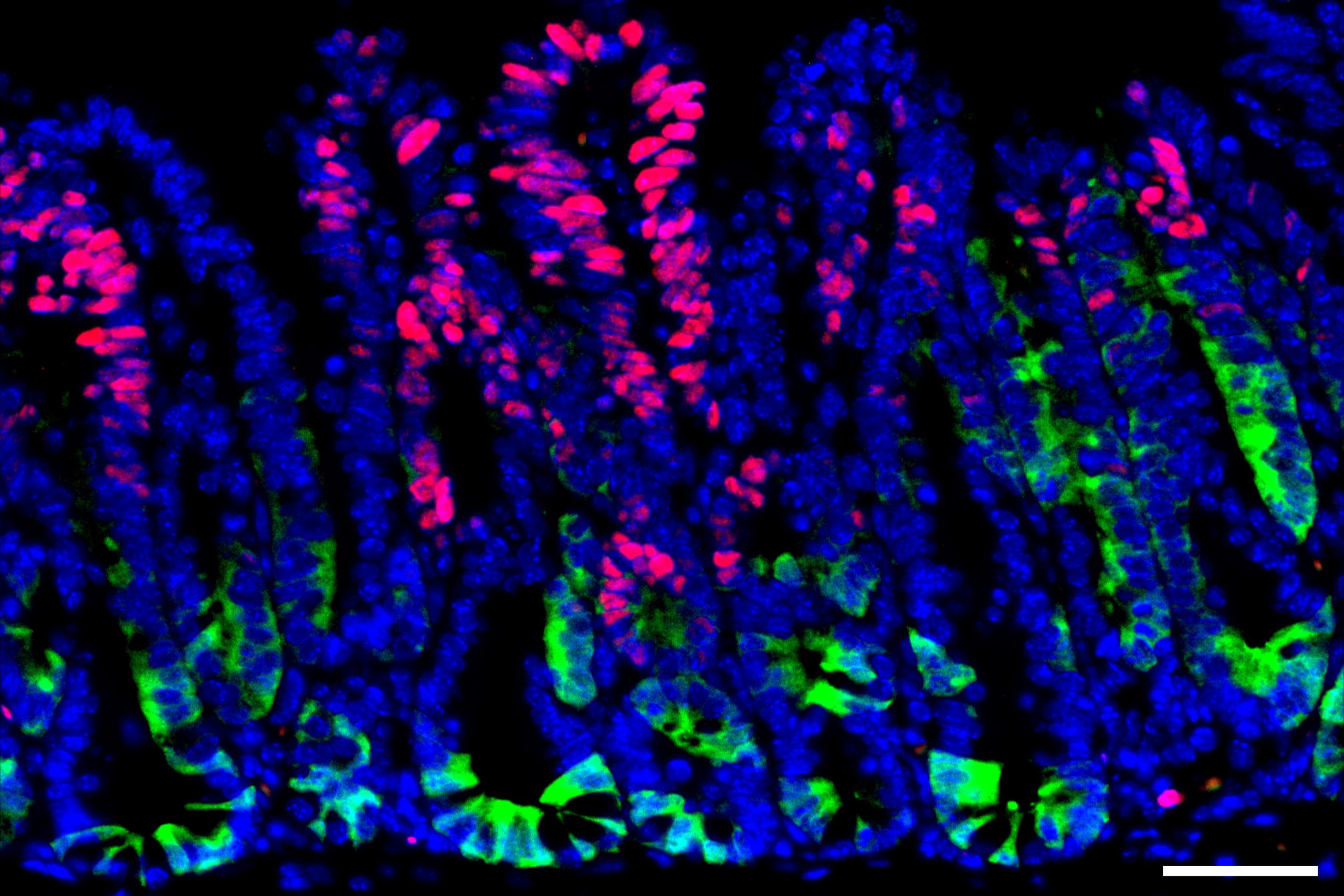
Fasting helps intestinal stem cells regenerate and heal injuries but also leads to a higher risk of cancer in mice, MIT researchers report.
Anne Trafton | MIT News
August 21, 2024
Low-calorie diets and intermittent fasting have been shown to have numerous health benefits: They can delay the onset of some age-related diseases and lengthen lifespan, not only in humans but many other organisms.
Many complex mechanisms underlie this phenomenon. Previous work from MIT has shown that one way fasting exerts its beneficial effects is by boosting the regenerative abilities of intestinal stem cells, which helps the intestine recover from injuries or inflammation.
In a study of mice, MIT researchers have now identified the pathway that enables this enhanced regeneration, which is activated once the mice begin “refeeding” after the fast. They also found a downside to this regeneration: When cancerous mutations occurred during the regenerative period, the mice were more likely to develop early-stage intestinal tumors.
“Having more stem cell activity is good for regeneration, but too much of a good thing over time can have less favorable consequences,” says Omer Yilmaz, an MIT associate professor of biology, a member of MIT’s Koch Institute for Integrative Cancer Research, and the senior author of the new study.
Yilmaz adds that further studies are needed before forming any conclusion as to whether fasting has a similar effect in humans.
“We still have a lot to learn, but it is interesting that being in either the state of fasting or refeeding when exposure to mutagen occurs can have a profound impact on the likelihood of developing a cancer in these well-defined mouse models,” he says.
MIT postdocs Shinya Imada and Saleh Khawaled are the lead authors of the paper, which appears today in Nature.
Driving regeneration
For several years, Yilmaz’s lab has been investigating how fasting and low-calorie diets affect intestinal health. In a 2018 study, his team reported that during a fast, intestinal stem cells begin to use lipids as an energy source, instead of carbohydrates. They also showed that fasting led to a significant boost in stem cells’ regenerative ability.
However, unanswered questions remained: How does fasting trigger this boost in regenerative ability, and when does the regeneration begin?
“Since that paper, we’ve really been focused on understanding what is it about fasting that drives regeneration,” Yilmaz says. “Is it fasting itself that’s driving regeneration, or eating after the fast?”
In their new study, the researchers found that stem cell regeneration is suppressed during fasting but then surges during the refeeding period. The researchers followed three groups of mice — one that fasted for 24 hours, another one that fasted for 24 hours and then was allowed to eat whatever they wanted during a 24-hour refeeding period, and a control group that ate whatever they wanted throughout the experiment.
The researchers analyzed intestinal stem cells’ ability to proliferate at different time points and found that the stem cells showed the highest levels of proliferation at the end of the 24-hour refeeding period. These cells were also more proliferative than intestinal stem cells from mice that had not fasted at all.
“We think that fasting and refeeding represent two distinct states,” Imada says. “In the fasted state, the ability of cells to use lipids and fatty acids as an energy source enables them to survive when nutrients are low. And then it’s the postfast refeeding state that really drives the regeneration. When nutrients become available, these stem cells and progenitor cells activate programs that enable them to build cellular mass and repopulate the intestinal lining.”
Further studies revealed that these cells activate a cellular signaling pathway known as mTOR, which is involved in cell growth and metabolism. One of mTOR’s roles is to regulate the translation of messenger RNA into protein, so when it’s activated, cells produce more protein. This protein synthesis is essential for stem cells to proliferate.
The researchers showed that mTOR activation in these stem cells also led to production of large quantities of polyamines — small molecules that help cells to grow and divide.
“In the refed state, you’ve got more proliferation, and you need to build cellular mass. That requires more protein, to build new cells, and those stem cells go on to build more differentiated cells or specialized intestinal cell types that line the intestine,” Khawaled says.
Too much of a good thing
The researchers also found that when stem cells are in this highly regenerative state, they are more prone to become cancerous. Intestinal stem cells are among the most actively dividing cells in the body, as they help the lining of the intestine completely turn over every five to 10 days. Because they divide so frequently, these stem cells are the most common source of precancerous cells in the intestine.
In this study, the researchers discovered that if they turned on a cancer-causing gene in the mice during the refeeding stage, they were much more likely to develop precancerous polyps than if the gene was turned on during the fasting state. Cancer-linked mutations that occurred during the refeeding state were also much more likely to produce polyps than mutations that occurred in mice that did not undergo the cycle of fasting and refeeding.
“I want to emphasize that this was all done in mice, using very well-defined cancer mutations. In humans it’s going to be a much more complex state,” Yilmaz says. “But it does lead us to the following notion: Fasting is very healthy, but if you’re unlucky and you’re refeeding after a fasting, and you get exposed to a mutagen, like a charred steak or something, you might actually be increasing your chances of developing a lesion that can go on to give rise to cancer.”
Yilmaz also noted that the regenerative benefits of fasting could be significant for people who undergo radiation treatment, which can damage the intestinal lining, or other types of intestinal injury. His lab is now studying whether polyamine supplements could help to stimulate this kind of regeneration, without the need to fast.
“This fascinating study provides insights into the complex interplay between food consumption, stem cell biology, and cancer risk,” says Ophir Klein, a professor of medicine at the University of California at San Francisco and Cedars-Sinai Medical Center, who was not involved in the study. “Their work lays a foundation for testing polyamines as compounds that may augment intestinal repair after injuries, and it suggests that careful consideration is needed when planning diet-based strategies for regeneration to avoid increasing cancer risk.”
The research was funded, in part, by a Pew-Stewart Trust Scholar award, the Marble Center for Cancer Nanomedicine, the Koch Institute-Dana Farber/Harvard Cancer Center Bridge Project, and the MIT Stem Cell Initiative.