Study suggests that stimulating stem cells may protect the gastrointestinal tract from age-related disease.
Anne Trafton | MIT News Office
March 28, 2019
Cells that line the intestinal tract are replaced every few days, a high rate of turnover that relies on a healthy population of intestinal stem cells. MIT and University of Tokyo biologists have now found that aging takes a toll on intestinal stem cells and may contribute to increased susceptibility to disorders of the gastrointestinal tract.
The researchers also showed that they could reverse this effect in aged mice by treating them with a compound that helps boost the population of intestinal stem cells. The findings suggest that this compound, which appears to stimulate a pathway that involves longevity-linked proteins known as sirtuins, could help protect the gut from age-related damage, the researchers say.
“One of the issues with aging is organ dysfunction, accompanied by a decline in the activity of the stem cells that nurture and replenish that organ, so this is a potentially very useful intervention point to either slow or reverse aging,” says Leonard Guarente, the Novartis Professor of Biology at MIT.
Guarente and Toshimasa Yamauchi, a professor at the University of Tokyo, are the senior authors of the study, which appears online in the journal Aging Cell on March 28. The lead author of the paper is Masaki Igarashi, a former MIT postdoc who is now at the University of Tokyo.
Population growth
Guarente’s lab has long studied the link between aging and sirtuins, a class of proteins found in nearly all animals. Sirtuins, which have been shown to protect against the effects of aging, can also be stimulated by calorie restriction.
In a paper published in 2016, Guarente and Igarashi found that in mice, low-calorie diets activate sirtuins in intestinal stem cells, helping the cells to proliferate. In their new study, they set out to investigate whether aging contributes to a decline in stem cell populations, and whether that decline could be reversed.
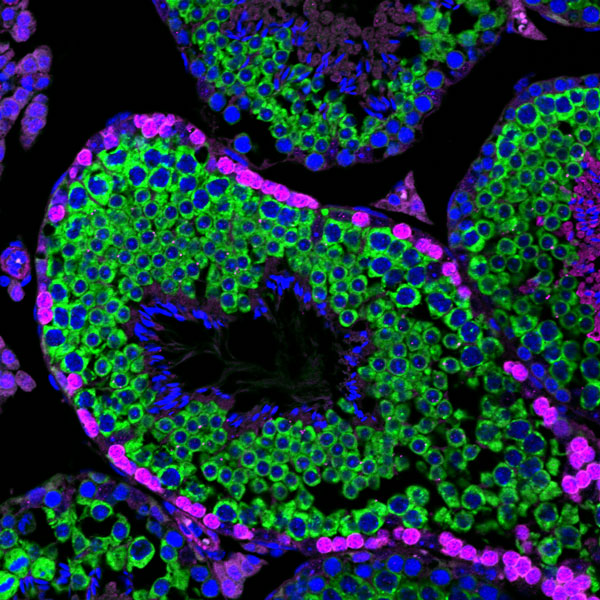
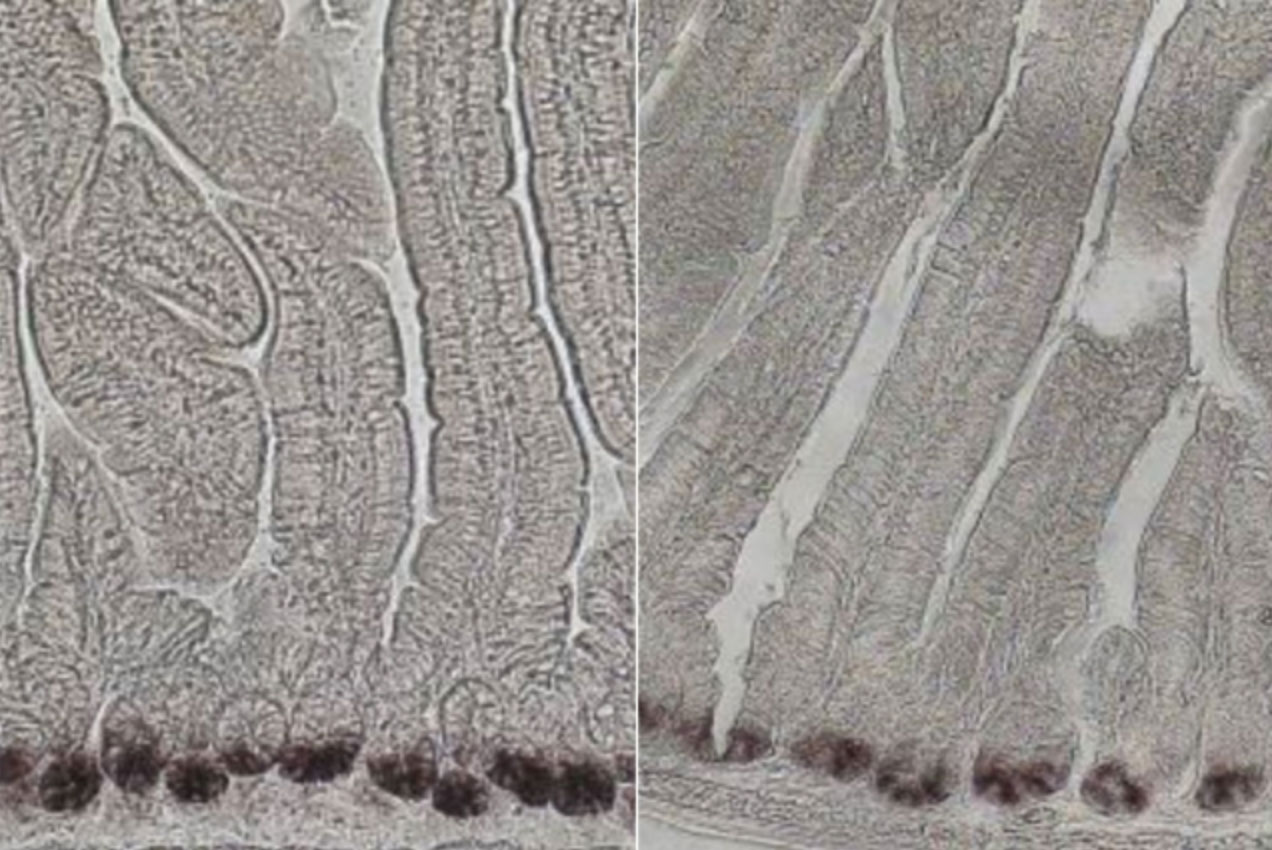
By comparing young (aged 3 to 5 months) and older (aged 2 years) mice, the researchers found that intestinal stem cell populations do decline with age. Furthermore, when these stem cells are removed from the mice and grown in a culture dish, they are less able to generate intestinal organoids, which mimic the structure of the intestinal lining, compared to stem cells from younger mice. The researchers also found reduced sirtuin levels in stem cells from the older mice.
Once the effects of aging were established, the researchers wanted to see if they could reverse the effects using a compound called nicotinamide riboside (NR). This compound is a precursor to NAD, a coenzyme that activates the sirtuin SIRT1. They found that after six weeks of drinking water spiked with NR, the older mice had normal levels of intestinal stem cells, and these cells were able to generate organoids as well as stem cells from younger mice could.
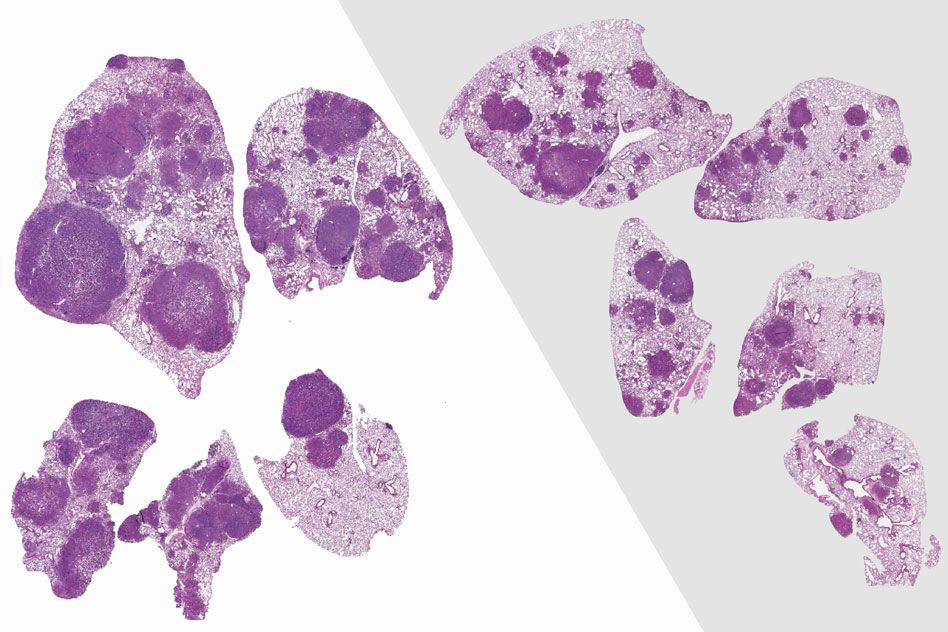
To determine if this stem cell boost actually has any health benefits, the researchers gave the older, NR-treated mice a compound that normally induces colitis. They found that NR protected the mice from the inflammation and tissue damage usually produced by this compound in older animals.
“That has real implications for health because just having more stem cells is all well and good, but it might not equate to anything in the real world,” Guarente says. “Knowing that the guts are actually more stress-resistant if they’re NR- supplemented is pretty interesting.”
Protective effects
Guarente says he believes that NR is likely acting through a pathway that his lab previously identified, in which boosting NAD turns on not only SIRT1 but another gene called mTORC1, which stimulates protein synthesis in cells and helps them to proliferate.
“What we would hypothesize is that the NAD replenishment in old mice is driving this pathway of growth that’s working through SIRT1 and TOR to reverse the decline that has occurred with aging,” he says.
The findings suggest that NAD might have a protective effect against diseases of the gut, such as colitis, in older people, he says. Guarente and his colleagues have previously found that NAD precursors can also stimulate the growth of blood vessels and muscles and boost endurance in aged mice, and a 2016 study from researchers in Switzerland found that boosting NAD can help replenish muscle stem cell populations in aged mice.
In 2014, Guarente started a company called Elysium Health, which sells a dietary supplement containing NR combined with another natural compound called pterostilbene, which is an activator of SIRT1.
The research was funded, in part, by the National Institutes of Health and the Glenn Foundation for Medical Research.