Biologists uncover an evolutionary trick to control gene expression that reverses the flow of genetic information from RNA splicing back to transcription.
Raleigh McElvery | Department of Biology
December 9, 2019
Sometimes, unexpected research results are simply due to experimental error. Other times, it’s the opposite — the scientists have uncovered a new phenomenon that reveals an even more accurate portrayal of our bodies and our universe, overturning well-established assumptions. Indeed, many great biological discoveries are made when results defy expectation.
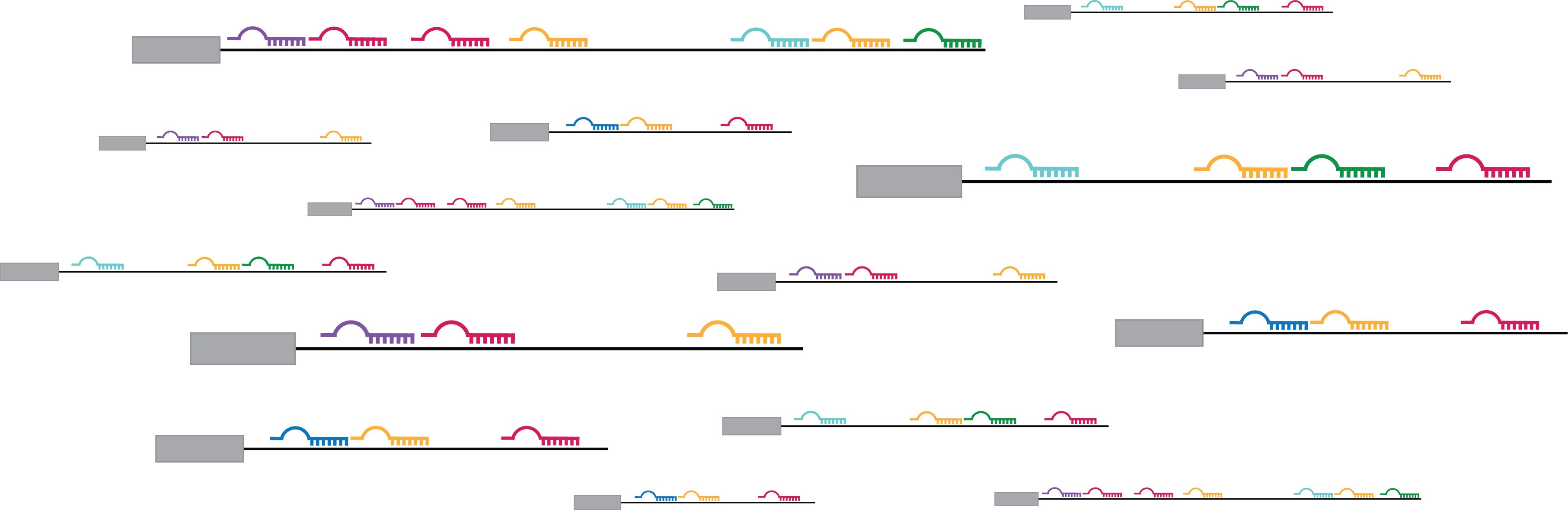
A few years ago, researchers in the Burge lab were comparing the genomic evolution of several different mammals when they noticed a strange pattern. Whenever a new nucleotide sequence appeared in the RNA of one lineage, there was generally an increase in the total amount of RNA produced from the gene in that lineage. Now, in a new paper, the Burge lab finally has an explanation, which redefines our understanding of how genes are expressed.
Once DNA is transcribed into RNA, the RNA transcript must be processed before it can be translated into proteins or go on to serve other roles within the cell. One important component of this processing is splicing, during which certain nucleotide sequences (introns) are removed from the newlymade RNA transcript, while others (the exons) remain. Depending on how the RNA is spliced, a single gene can give rise to a diverse array of transcripts.
Given this order of operations, it makes sense that transcription affects splicing. After all, splicing cannot occur without an RNA transcript. But the inverse theory — that splicing can affect transcription — is now gaining traction. In a recent study, the Burge lab showed that splicing in an exon near the beginning of a gene impacts transcription and increases gene expression, offering an explanation for the patterns in their previous findings.
“Rather than Step A impacting Step B, what we found here is that Step B, splicing, actually feeds back to influence Step A, transcription,” says Christopher Burge, senior author and professor of biology. “It seems contradictory, since splicing requires transcription, but there is actually no contradiction if — as in our model — the splicing of one transcript from a gene influences the transcription of subsequent transcripts from the same gene.”
The study, published on Nov. 28 in Cell, was led by Burge lab postdoc Ana Fiszbein.
Promoting gene expression
In order for transcription to begin, molecular machines must be recruited to a special sequence of DNA, known as the promoter. Some promoters are better at recruiting this machinery than others, and therefore initiate transcription more often. However, having different promoters available to produce slightly different transcripts from a gene helps boost expression and generates transcript diversity, even before splicing occurs mere seconds or minutes later.
At first, Fiszbein wasn’t sure how the new exons were enhancing gene expression, but she theorized that new promoters were involved. Based on evolutionary data available and her experiments at the lab bench, she could see that wherever there was a new exon, there was usually a new promoter nearby. When the exon was spliced in, the new promoter became more active.
The researchers named this phenomenon “exon-mediated activation of transcription starts” (EMATS). They propose a model in which the splicing machinery associated with the new exon recruits transcription machinery to the vicinity, activating transcription from nearby promoters. This process, the researchers predict, likely helps to regulate thousands of mammalian genes across species.
A more flexible genome
Fiszbein believes that EMATS has increased genome complexity over the course of evolution, and may have contributed to species-specific differences. For instance, the mouse and rat genomes are quite similar, but EMATS could have helped produce new promoters, leading to regulatory changes that drive differences in structure and function between the two. EMATS may also contribute to differences in expression between tissues in the same organism.
“EMATS adds a new layer of complexity to gene expression regulation,” Fiszbein says. “It gives the genome more flexibility, and introduces the potential to alter the amount of RNA produced.”
Juan Valcárcel, a research professor at the Catalan Institution for Research and Advanced Studies in the Center for Genomic Regulation in Barcelona, Spain, says understanding the mechanisms behind EMATS could also have biotechnological and therapeutic implications. “A number of human conditions, including genetic diseases and cancer, are caused by a defect or an excess of particular genes,” he says. “Reverting these anomalies through modulation of EMATS might provide innovative therapies.”
Researchers have already begun to tinker with splicing to control transcription. According to Burge, pharmaceutical companies like Ionis, Novartis, and Roche are concocting drugs to regulate splicing and treat diseases like spinal muscular atrophy. There are many ways to decrease gene expression, but it’s much harder to increase it in a targeted manner. “Tweaking splicing might be one way to do that,” he says.
“We found a way in which our cells change gene expression,” Fiszbein adds. “And we can use that to manipulate transcript levels as we want. I think that’s the most exciting part.”
This research was funded by the National Institutes of Health and the Pew Latin American Fellows Program in the Biomedical Sciences.