Graduate student Ellen Zhong helped biologists and mathematicians reach across departmental lines to address a longstanding problem in electron microscopy.
Saima Sidik | Department of Biology
July 1, 2021
MIT’s Hockfield Court is bordered on the west by the ultramodern Stata Center, with its reflective, silver alcoves that jut off at odd angles, and on the east by Building 68, which is a simple, window-lined, cement rectangle. At first glance, Bonnie Berger’s mathematics lab in the Stata Center and Joey Davis’s biology lab in Building 68 are as different as the buildings that house them. And yet, a recent collaboration between these two labs shows how their disciplines complement each other. The partnership started when Ellen Zhong, a graduate student from the Computational and Systems Biology (CSB) Program, decided to use a computational pattern-recognition tool called a neural network to study the shapes of molecular machines. Three years later, Zhong’s project is letting scientists see patterns that run beneath the surface of their data, and deepening their understanding of the molecules that shape life.
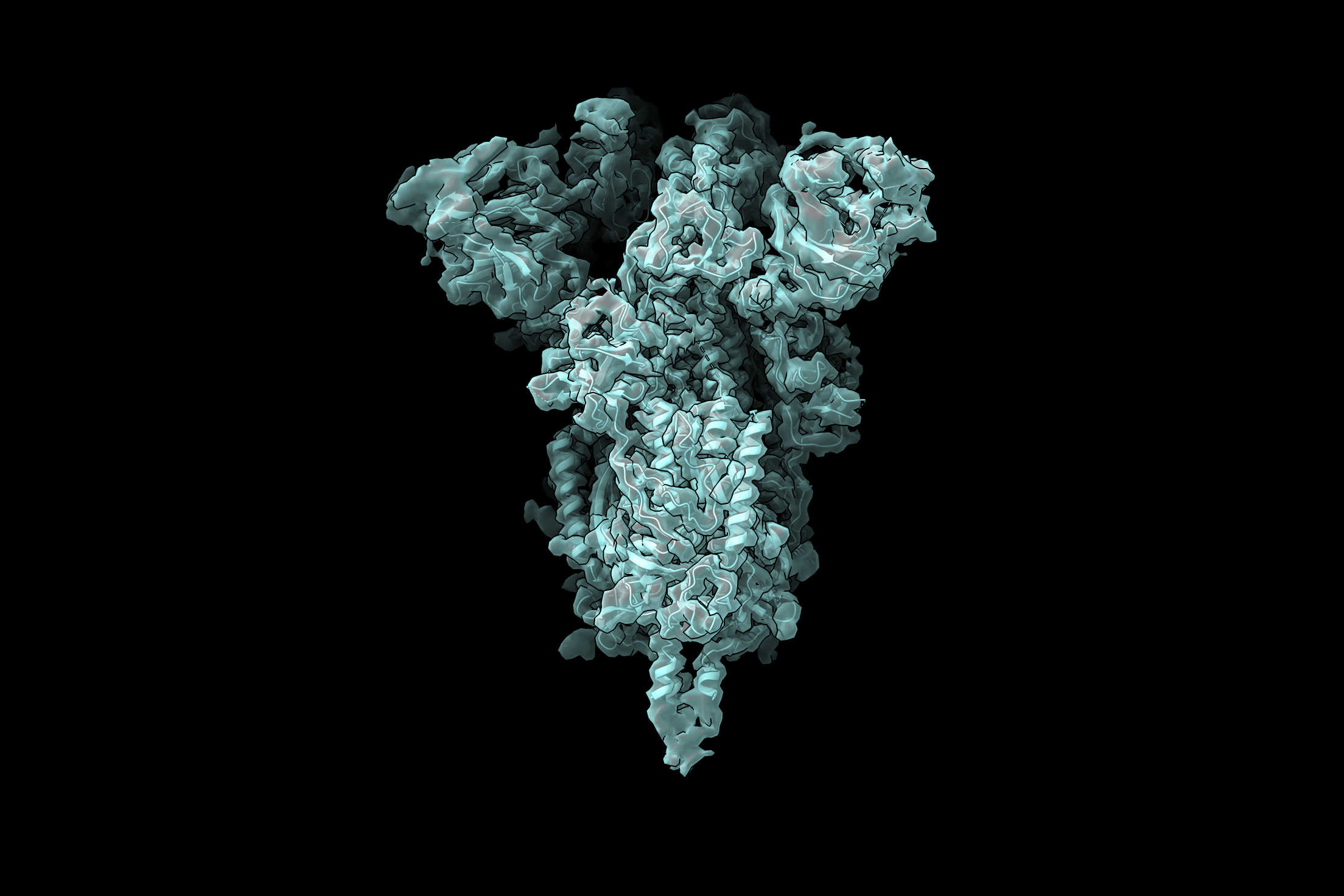
Zhong’s work builds on a technique from the 1970s called cryo-electron microscopy (cryo-EM), which lets researchers take high-resolution images of frozen protein complexes. Over the past decade, better microscopes and cameras have led to a “resolution revolution” in cryo-EM that’s allowed scientists to see individual atoms within proteins. But, as good as these images are, they’re still only static snapshots. In reality, many of these molecular machines are constantly changing shape and composition as cells carry out their normal functions and adjust to new situations.
Along with former Berger lab member Tristan Belper, Zhong devised software called cryoDRGN. The tool uses neural nets to combine hundreds of thousands of cryo-EM images, and shows scientists the full range of three-dimensional conformations that protein complexes can take, letting them reconstruct the proteins’ motion as they carry out cellular functions. Understanding the range of shapes that protein complexes can take helps scientists develop drugs that block viruses from entering cells, study how pests kill crops, and even design custom proteins that can cure disease. Covid-19 vaccines, for example, work partly because they include a mutated version of the virus’s spike protein that’s stuck in its active conformation, so vaccinated people produce antibodies that block the virus from entering human cells. Scientists needed to understand the variety of shapes that spike proteins can take in order to figure out how to force spike into its active conformation.
Getting off the computer and into the lab
Zhong’s interest in computational biology goes back to 2011 when, as a chemical engineering undergrad at the University of Virginia, she worked with Professor Michael Shirts to simulate how proteins fold and unfold. After college, Zhong took her skills to a company called D. E. Shaw Research, where, as a scientific programmer, she took a computational approach to studying how proteins interact with small-molecule drugs.
“The research was very exciting,” Zhong says, “but all based on computer simulations. To really understand biological systems, you need to do experiments.”
This goal of combining computation with experimentation motivated Zhong to join MIT’s CSB PhD program, where students often work with multiple supervisors to blend computational work with bench work. Zhong “rotated” in both the Davis and Berger labs, then decided to combine the Davis lab’s goal of understanding how protein complexes form with the Berger lab’s expertise in machine learning and algorithms. Davis was interested in building up the computational side of his lab, so he welcomed the opportunity to co-supervise a student with Berger, who has a long history of collaborating with biologists.
Davis himself holds a dual bachelor’s degree in computer science and biological engineering, so he’s long believed in the power of combining complementary disciplines. “There are a lot of things you can learn about biology by looking in a microscope,” he says. “But as we start to ask more complicated questions about entire systems, we’re going to require computation to manage the high-dimensional data that come back.”
Before rotating in the Davis lab, Zhong had never performed bench work before — or even touched a pipette. She was fascinated to find how streamlined some very powerful molecular biology techniques can be. Still, Zhong realized that physical limitations mean that biology is much slower when it’s done at the bench instead of on a computer. “With computational research, you can automate experiments and run them super quickly, whereas in the wet lab, you only have two hands, so you can only do one experiment at a time,” she says.
Zhong says that synergizing the two different cultures of the Davis and Berger labs is helping her become a well-rounded, adaptable scientist. Working around experimentalists in the Davis lab has shown her how much labor goes into experimental results, and also helped her to understand the hurdles that scientists face at the bench. In the Berger lab, she enjoys having coworkers who understand the challenges of computer programming.
“The key challenge in collaborating across disciplines is understanding each other’s ‘languages,’” Berger says. “Students like Ellen are fortunate to be learning both biology and computing dialects simultaneously.”
Bringing in the community
Last spring revealed another reason for biologists to learn computational skills: these tools can be used anywhere there’s a computer and an internet connection. When the Covid-19 pandemic hit, Zhong’s colleagues in the Davis lab had to wind down their bench work for a few months, and many of them filled their time at home by using cryo-EM data that’s freely available online to help Zhong test her cryoDRGN software. The difficulty of understanding another discipline’s language quickly became apparent, and Zhong spent a lot of time teaching her colleagues to be programmers. Seeing the problems that nonprogrammers ran into when they used cryoDRGN was very informative, Zhong says, and helped her create a more user-friendly interface.
Although the paper announcing cryoDRGN was just published in February, the tool created a stir as soon as Zhong posted her code online, many months prior. The cryoDRGN team thinks this is because leveraging knowledge from two disciplines let them visualize the full range of structures that protein complexes can have, and that’s something researchers have wanted to do for a long time. For example, the cryoDRGN team recently collaborated with researchers from Harvard and Washington universities to study locomotion of the single-celled organism Chlamydomonas reinhardtii. The mechanisms they uncovered could shed light on human health conditions, like male infertility, that arise when cells lose the ability to move. The team is also using cryoDRGN to study the structure of the SARS-CoV-2 spike protein, which could help scientists design treatments and vaccines to fight coronaviruses.
Zhong, Berger, and Davis say they’re excited to continue using neural nets to improve cryo-EM analysis, and to extend their computational work to other aspects of biology. Davis cited mass spectrometry as “a ripe area to apply computation.” This technique can complement cryo-EM by showing researchers the identities of proteins, how many of them are bound together, and how cells have modified them.
“Collaborations between disciplines are the future,” Berger says. “Researchers focused on a single discipline can take it only so far with existing techniques. Shining a different lens on the problem is how advances can be made.”
Zhong says it’s not a bad way to spend a PhD, either. Asked what she’d say to incoming graduate students considering interdisciplinary projects, she says: “Definitely do it.”