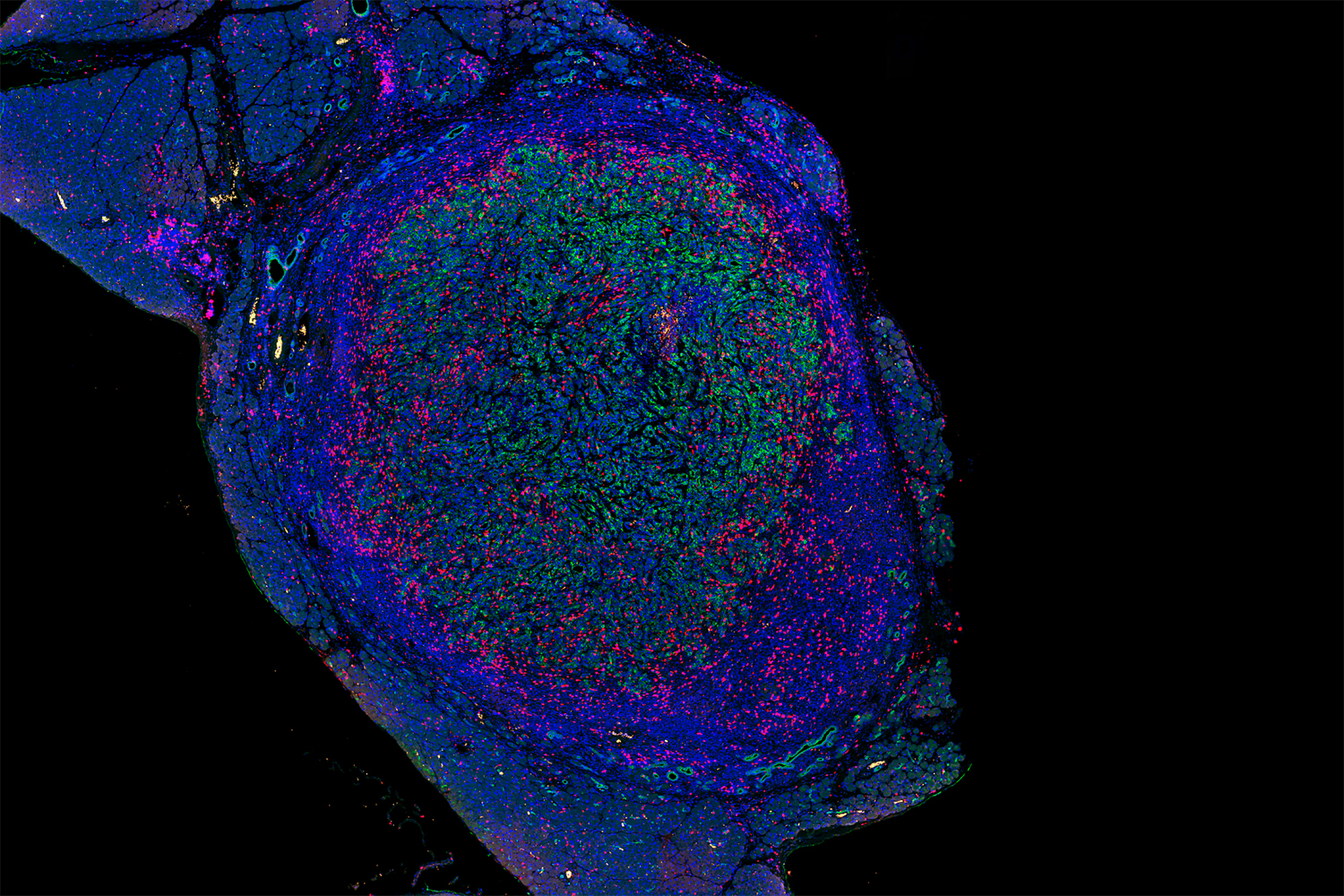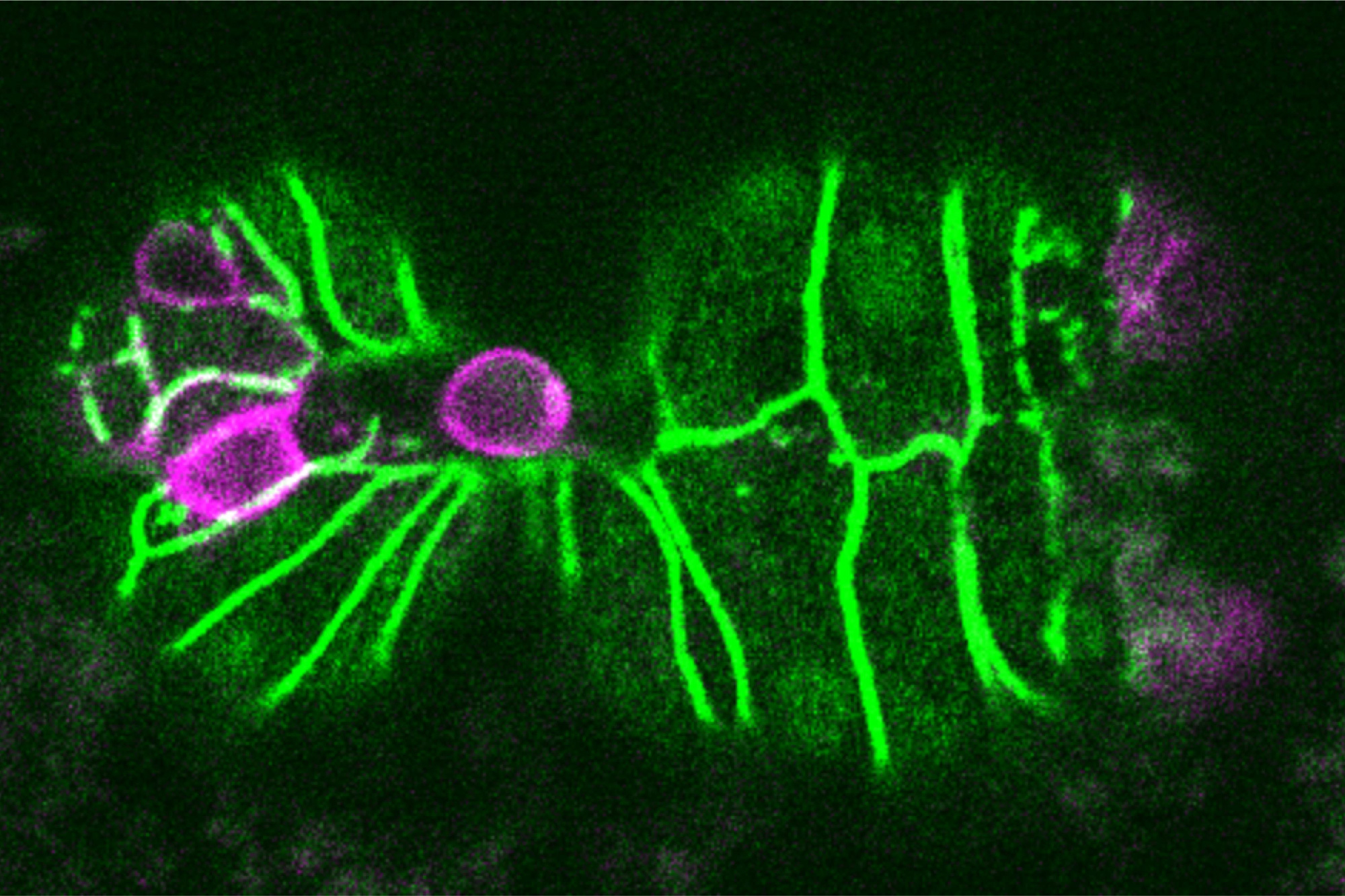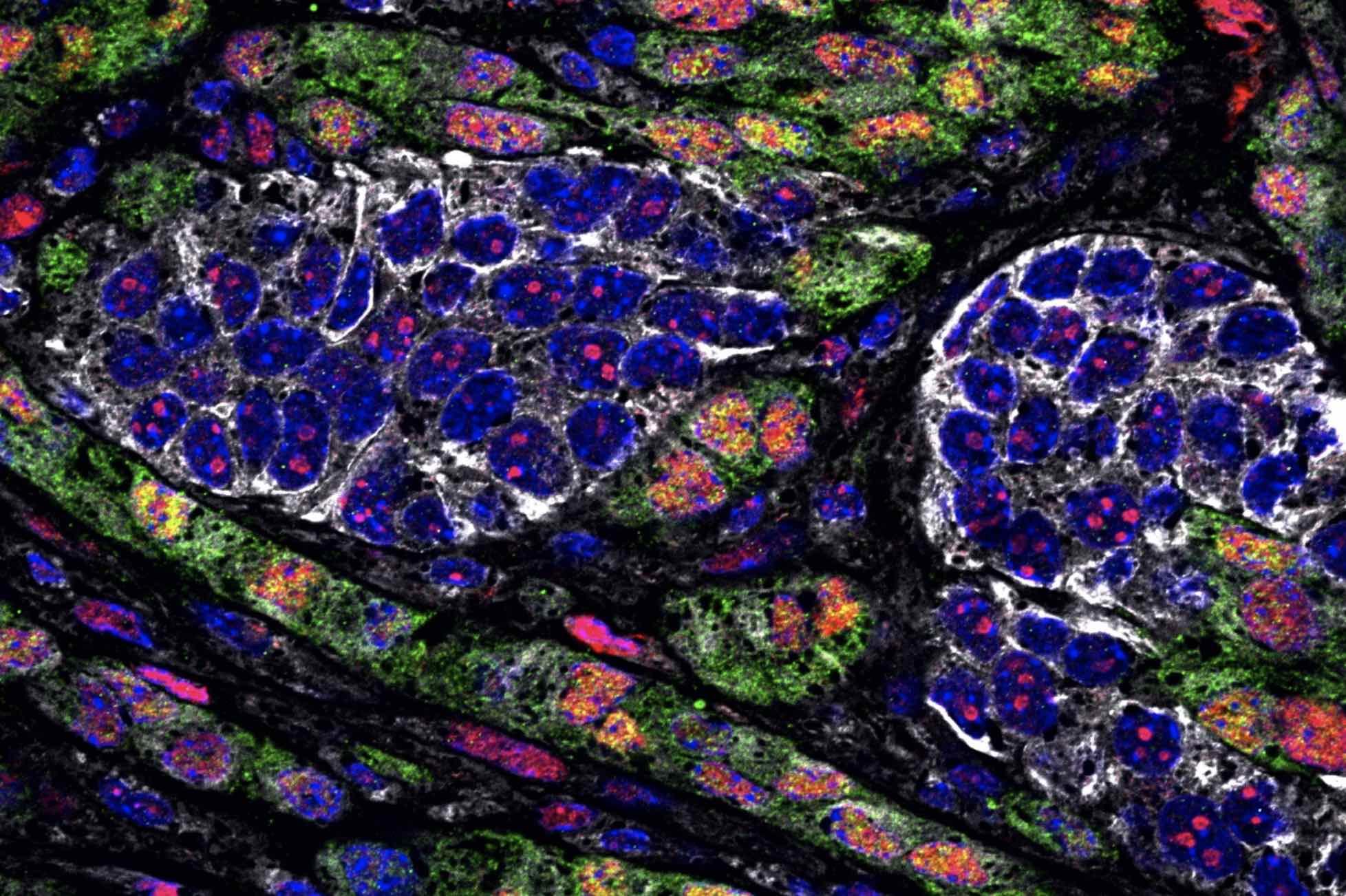
Vaccinating against certain proteins found on cancer cells could help to enhance the T cell response to tumors.
Anne Trafton | MIT News Office
September 16, 2021
Over the past decade, scientists have been exploring vaccination as a way to help fight cancer. These experimental cancer vaccines are designed to stimulate the body’s own immune system to destroy a tumor, by injecting fragments of cancer proteins found on the tumor.
So far, none of these vaccines have been approved by the FDA, but some have shown promise in clinical trials to treat melanoma and some types of lung cancer. In a new finding that may help researchers decide what proteins to include in cancer vaccines, MIT researchers have found that vaccinating against certain cancer proteins can boost the overall T cell response and help to shrink tumors in mice.
The research team found that vaccinating against the types of proteins they identified can help to reawaken dormant T cell populations that target those proteins, strengthening the overall immune response.
“This study highlights the importance of exploring the details of immune responses against cancer deeply. We can now see that not all anticancer immune responses are created equal, and that vaccination can unleash a potent response against a target that was otherwise effectively ignored,” says Tyler Jacks, the David H. Koch Professor of Biology, a member of the Koch Institute for Integrative Cancer Research, and the senior author of the study.
MIT postdoc Megan Burger is the lead author of the new study, which appears today in Cell.
T cell competition
When cells begin to turn cancerous, they start producing mutated proteins not seen in healthy cells. These cancerous proteins, also called neoantigens, can alert the body’s immune system that something has gone wrong, and T cells that recognize those neoantigens start destroying the cancerous cells.
Eventually, these T cells experience a phenomenon known as “T cell exhaustion,” which occurs when the tumor creates an immunosuppressive environment that disables the T cells, allowing the tumor to grow unchecked.
Scientists hope that cancer vaccines could help to rejuvenate those T cells and help them to attack tumors. In recent years, they have worked to develop methods for identifying neoantigens in patient tumors to incorporate into personalized cancer vaccines. Some of these vaccines have shown promise in clinical trials to treat melanoma and non-small cell lung cancer.
“These therapies work amazingly in a subset of patients, but the vast majority still don’t respond very well,” Burger says. “A lot of the research in our lab is aimed at trying to understand why that is and what we can do therapeutically to get more of those patients responding.”
Previous studies have shown that of the hundreds of neoantigens found in most tumors, only a small number generate a T cell response.
The new MIT study helps to shed light on why that is. In studies of mice with lung tumors, the researchers found that as tumor-targeting T cells arise, subsets of T cells that target different cancerous proteins compete with each other, eventually leading to the emergence of one dominant population of T cells. After these T cells become exhausted, they still remain in the environment and suppress any competing T cell populations that target different proteins found on the tumor.
However, Burger found that if she vaccinated these mice with one of the neoantigens targeted by the suppressed T cells, she could rejuvenate those T cell populations.
“If you vaccinate against antigens that have suppressed responses, you can unleash those T cell responses,” she says. “Trying to identify these suppressed responses and specifically targeting them might improve patient responses to vaccine therapies.”
Shrinking tumors
In this study, the researchers found that they had the most success when vaccinating with neoantigens that bind weakly to immune cells that are responsible for presenting the antigen to T cells. When they used one of those neoantigens to vaccinate mice with lung tumors, they found the tumors shrank by an average of 27 percent.
“The T cells proliferate more, they target the tumors better, and we see an overall decrease in lung tumor burden in our mouse model as a result of the therapy,” Burger says.
After vaccination, the T cell population included a type of cells that have the potential to continuously refuel the response, which could allow for long-term control of a tumor.
In future work, the researchers hope to test therapeutic approaches that would combine this vaccination strategy with cancer drugs called checkpoint inhibitors, which can take the brakes off exhausted T cells, stimulating them to attack tumors. Supporting that approach, the results published today also indicate that vaccination boosts the number of a specific type of T cells that have been shown to respond well to checkpoint therapies.
The research was funded by the Howard Hughes Medical Institute, the Ludwig Center at Harvard University, the National Institutes of Health, the Koch Institute Support (core) Grant from the National Cancer Institute, the Bridge Project of the Koch Institute and Dana-Farber/Harvard Cancer Center, and fellowship awards from the Jane Coffin Childs Memorial Fund for Medical Research and the Ludwig Center for Molecular Oncology at MIT.