Stefani Spranger is working to discover why some cancers don’t respond to immunotherapy, in hopes of making them more vulnerable to it.
Anne Trafton | MIT News
February 26, 2025
In addition to patrolling the body for foreign invaders, the immune system also hunts down and destroys cells that have become cancerous or precancerous. However, some cancer cells end up evading this surveillance and growing into tumors.
Once established, tumor cells often send out immunosuppressive signals, which leads T cells to become “exhausted” and unable to attack the tumor. In recent years, some cancer immunotherapy drugs have shown great success in rejuvenating those T cells so they can begin attacking tumors again.
While this approach has proven effective against cancers such as melanoma, it doesn’t work as well for others, including lung and ovarian cancer. MIT Associate Professor Stefani Spranger is trying to figure out how those tumors are able to suppress immune responses, in hopes of finding new ways to galvanize T cells into attacking them.
“We really want to understand why our immune system fails to recognize cancer,” Spranger says. “And I’m most excited about the really hard-to-treat cancers because I think that’s where we can make the biggest leaps.”
Her work has led to a better understanding of the factors that control T-cell responses to tumors, and raised the possibility of improving those responses through vaccination or treatment with immune-stimulating molecules called cytokines.
“We’re working on understanding what exactly the problem is, and then collaborating with engineers to find a good solution,” she says.
Jumpstarting T cells
As a student in Germany, where students often have to choose their college major while still in high school, Spranger envisioned going into the pharmaceutical industry and chose to major in biology. At Ludwig Maximilian University in Munich, her course of study began with classical biology subjects such as botany and zoology, and she began to doubt her choice. But, once she began taking courses in cell biology and immunology, her interest was revived and she continued into a biology graduate program at the university.
During a paper discussion class early in her graduate school program, Spranger was assigned to a Science paper on a promising new immunotherapy treatment for melanoma. This strategy involves isolating tumor-infiltrating T-cells during surgery, growing them into large numbers, and then returning them to the patient. For more than 50 percent of those patients, the tumors were completely eliminated.
“To me, that changed the world,” Spranger recalls. “You can take the patient’s own immune system, not really do all that much to it, and then the cancer goes away.”
Spranger completed her PhD studies in a lab that worked on further developing that approach, known as adoptive T-cell transfer therapy. At that point, she still was leaning toward going into pharma, but after finishing her PhD in 2011, her husband, also a biologist, convinced her that they should both apply for postdoc positions in the United States.
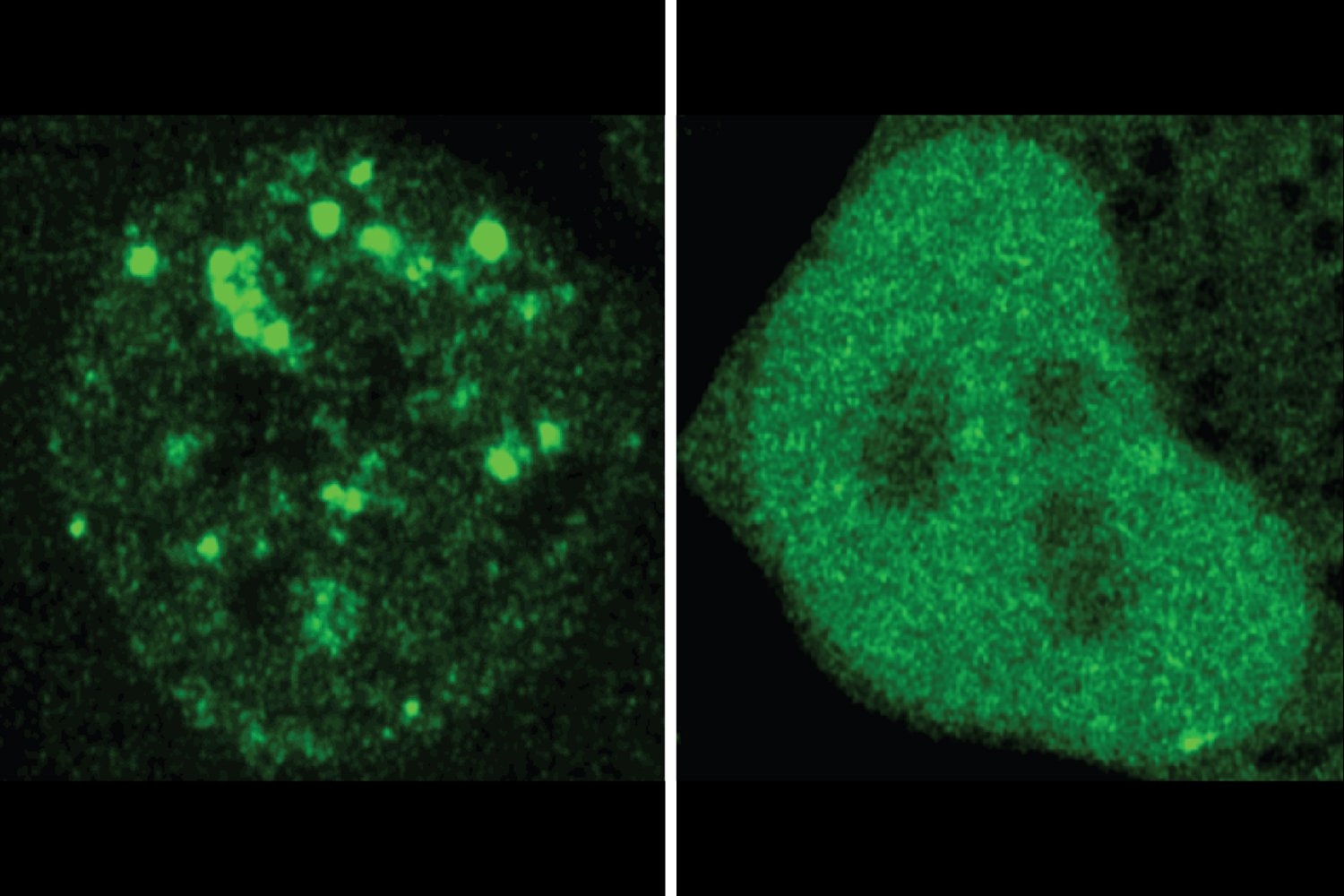
They ended up at the University of Chicago, where Spranger worked in a lab that studies how the immune system responds to tumors. There, she discovered that while melanoma is usually very responsive to immunotherapy, there is a small fraction of melanoma patients whose T cells don’t respond to the therapy at all. That got her interested in trying to figure out why the immune system doesn’t always respond to cancer the way that it should, and in finding ways to jumpstart it.
During her postdoc, Spranger also discovered that she enjoyed mentoring students, which she hadn’t done as a graduate student in Germany. That experience drew her away from going into the pharmaceutical industry, in favor of a career in academia.
“I had my first mentoring teaching experience having an undergrad in the lab, and seeing that person grow as a scientist, from barely asking questions to running full experiments and coming up with hypotheses, changed how I approached science and my view of what academia should be for,” she says.
Modeling the immune system
When applying for faculty jobs, Spranger was drawn to MIT by the collaborative environment of MIT and its Koch Institute for Integrative Cancer Research, which offered the chance to collaborate with a large community of engineers who work in the field of immunology.
“That community is so vibrant, and it’s amazing to be a part of it,” she says.
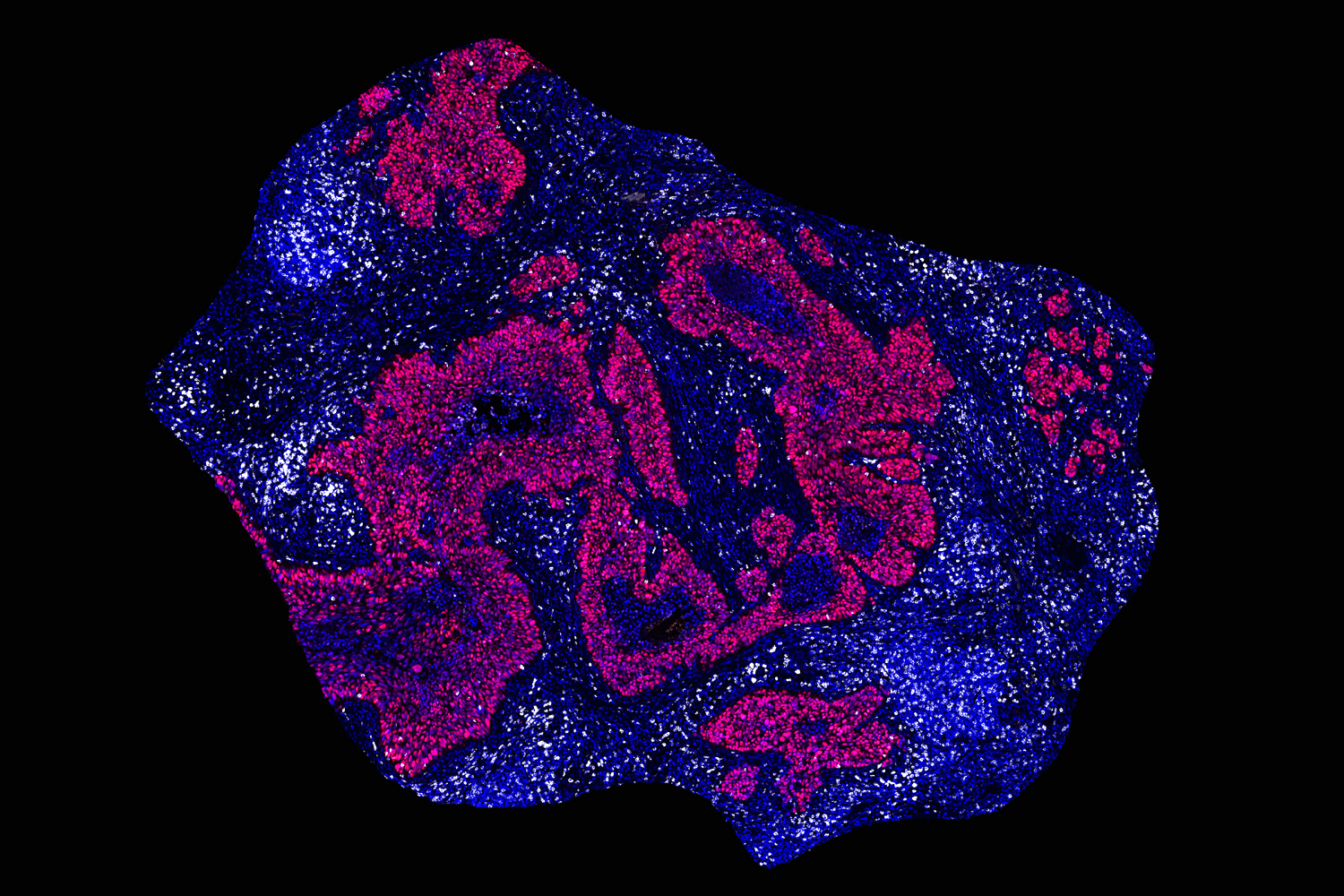
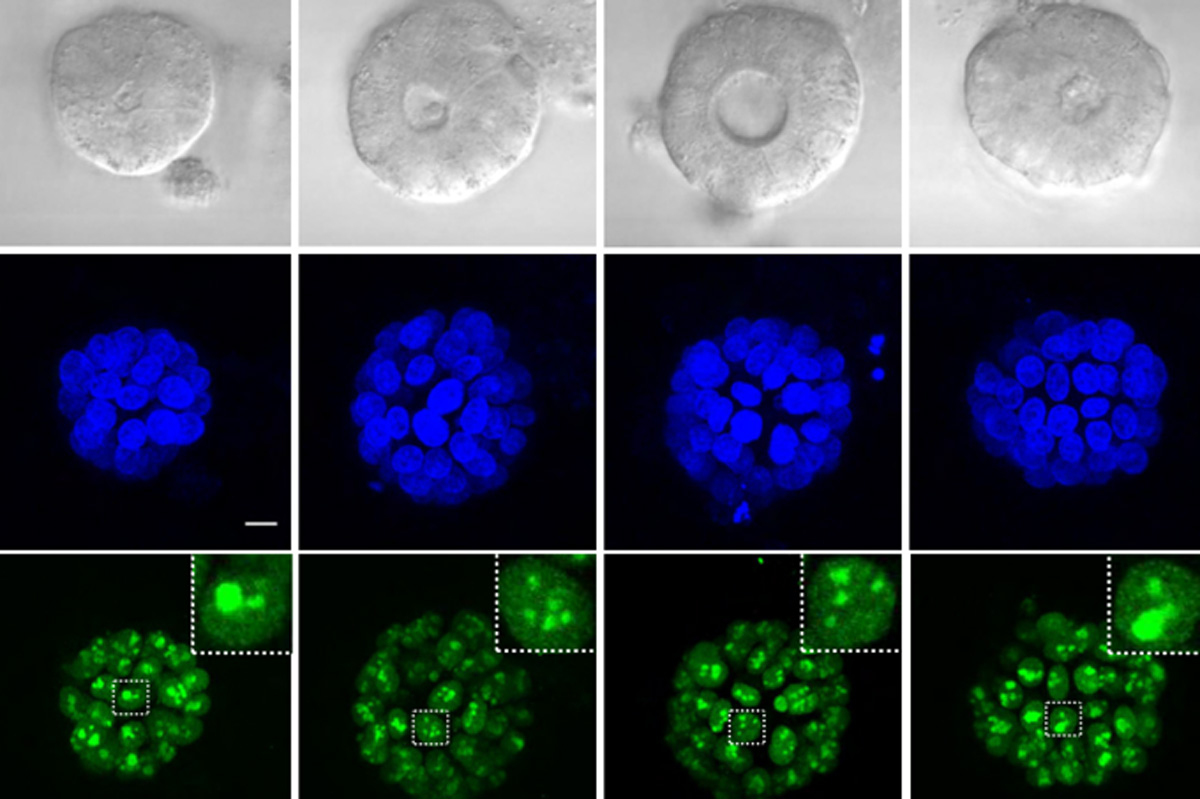
Building on the research she had done as a postdoc, Spranger wanted to explore why some tumors respond well to immunotherapy, while others do not. For many of her early studies, she used a mouse model of non-small-cell lung cancer. In human patients, the majority of these tumors do not respond well to immunotherapy.
“We build model systems that resemble each of the different subsets of non-responsive non-small cell lung cancer, and we’re trying to really drill down to the mechanism of why the immune system is not appropriately responding,” she says.
As part of that work, she has investigated why the immune system behaves differently in different types of tissue. While immunotherapy drugs called checkpoint inhibitors can stimulate a strong T-cell response in the skin, they don’t do nearly as much in the lung. However, Spranger has shown that T cell responses in the lung can be improved when immune molecules called cytokines are also given along with the checkpoint inhibitor.
Those cytokines work, in part, by activating dendritic cells — a class of immune cells that help to initiate immune responses, including activation of T cells.
“Dendritic cells are the conductor for the orchestra of all the T cells, although they’re a very sparse cell population,” Spranger says. “They can communicate which type of danger they sense from stressed cells and then instruct the T cells on what they have to do and where they have to go.”
Spranger’s lab is now beginning to study other types of tumors that don’t respond at all to immunotherapy, including ovarian cancer and glioblastoma. Both the brain and the peritoneal cavity appear to suppress T-cell responses to tumors, and Spranger hopes to figure out how to overcome that immunosuppression.
“We’re specifically focusing on ovarian cancer and glioblastoma, because nothing’s working right now for those cancers,” she says. “We want to understand what we have to do in those sites to induce a really good anti-tumor immune response.”