Eliezer Calo’s studies of craniofacial malformations have yielded insight into protein synthesis and embryonic development.
Anne Trafton | MIT News
March 6, 2026
When Associate Professor Eliezer Calo PhD ’11 was applying for faculty positions, he was drawn to MIT not only because it’s his alma mater, but also because the Department of Biology places high value on exploring fundamental questions in biology.
In his own lab, Calo studies how craniofacial malformations arise. One motivation is to seek new treatments for those conditions, but another is to learn more about fundamental biological processes such as protein synthesis and embryonic development.
“We use genes that are mutated in disease to uncover fundamental biology,” Calo says. “Mutations that happen in disease are an experiment of nature, telling us that those are the important genes, and then we follow them up not only to understand the disease, but to fundamentally understand what the genes are doing.”
Calo’s work has led to new insights into how ribosomes form and how they control protein synthesis, as well as how the nucleolus, the birthplace of ribosomes in eukaryotic cells, has evolved over hundreds of millions of years.
In addition to earning his PhD at MIT, Calo is also an alumnus of MIT’s Summer Research Program (MSRP), which helps to prepare undergraduate students to pursue graduate education. Since starting his lab at MIT, Calo has made a point to serve as a research mentor for the program every summer.
“I feel that it’s important to pay back to the program that helped me realize what I wanted to do,” he says.
A nontraditional path
Growing up in a mountainous region of Puerto Rico, Calo was the first person from his family to finish high school. While attending the University of Puerto Rico at Rio Piedras, the largest university in Puerto Rico, he explored a few different majors before settling on chemistry.
One of Calo’s chemistry professors invited him to work in her lab, where he did a research project studying the pharmacokinetics of cell receptors found on the surface of astrocytes, a type of brain cell.
“It was a good mix of biology and chemistry,” he says. “I think that that was the catalyst to my pursuit of a career in the sciences.”
He learned about MSRP from Mandana Sassanfar, a senior lecturer in biology at MIT and director of outreach for several MIT departments, at an event hosted by the University of Puerto Rico for students interested in careers in science. He was accepted into the program, and during the summer after his junior year, he worked in the lab of Stephen Bell, an MIT professor of biology. That experience, he says, was transformative.
“Without that experience, I would have probably chosen another career,” Calo says. In Puerto Rico, “science was fun, but it was a struggle. We had to make everything from scratch, and then you spend more time making reagents than doing the experiments. When I came to MIT, I was always doing experiments.”
During that time, he realized he liked working in biology labs more than chemistry labs, so when he applied to graduate school, he decided to move into biology. He applied to five schools, including MIT. “Once MIT sent me the acceptance, I just had to say yes. There was no saying no.”
At MIT, Calo thought he might study biochemistry, but he ended up focusing on cancer biology instead, working with Jacqueline Lees, an MIT biology professor, to study the role of the tumor suppressor protein Rb.
After finishing his PhD, Calo felt burnt out and wasn’t sure if he wanted to continue along the academic track. His thesis committee advisors encouraged him to do a postdoc just to try it out, and he ended up going to Stanford University, where he fell in love with California and switched to a new research focus. Working with Joanna Wysocka, a professor of developmental biology at Stanford, he began investigating how development is affected by the regulation of proteins that make up cellular ribosomes — a topic his lab still studies today.
Returning to MIT
When searching for faculty jobs, Calo focused mainly on schools in California, but also sent an application to MIT. As he was deciding between offers from MIT and the University of California at Berkeley, a phone call from Angelika Amon, the late MIT professor of biology, convinced him to take the cross-country leap back to MIT.
“She had me on the phone for more than one hour telling me why I should come to MIT,” he recalls. “And that was so heartwarming that I could not say no.”
Since starting his lab in 2017, Calo has been studying how defects in the production of ribosomes give rise to diseases, in particular craniofacial malformations such as cleft palate.
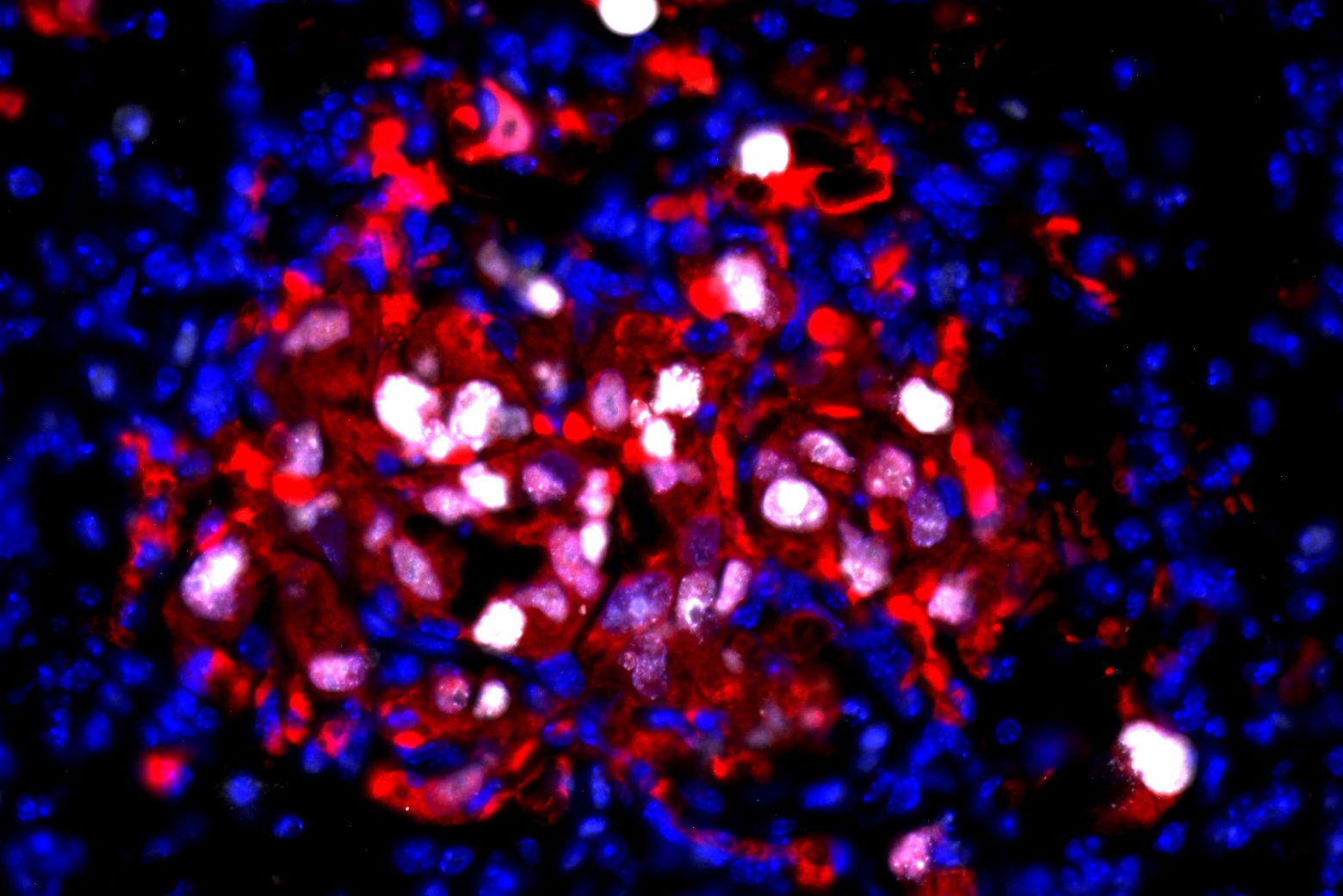
Ribosomes, the organelles where protein synthesis occurs, consist of two subunits made of about 80 proteins. A longstanding question in biology has been why mutations that affect ribosome formation appear to primarily affect the development of the face, but not the rest of the body.
In a 2018 study, Calo discovered that this is because the mutations that affect ribosomes can have secondary effects that influence craniofacial development. In embryonic cells that form the face, a mutation in a gene called TCOF1 activates p53 at a higher level than in other embryonic cells. High levels of p53 cause some of those cells to undergo programmed cell death, leading to Treacher-Collins Syndrome, a disorder that produces underdeveloped bones in the jaw and cheek.
His lab has shown that p53 overactivation is also responsible for craniofacial disorders caused by mutations in RNA splicing factors.
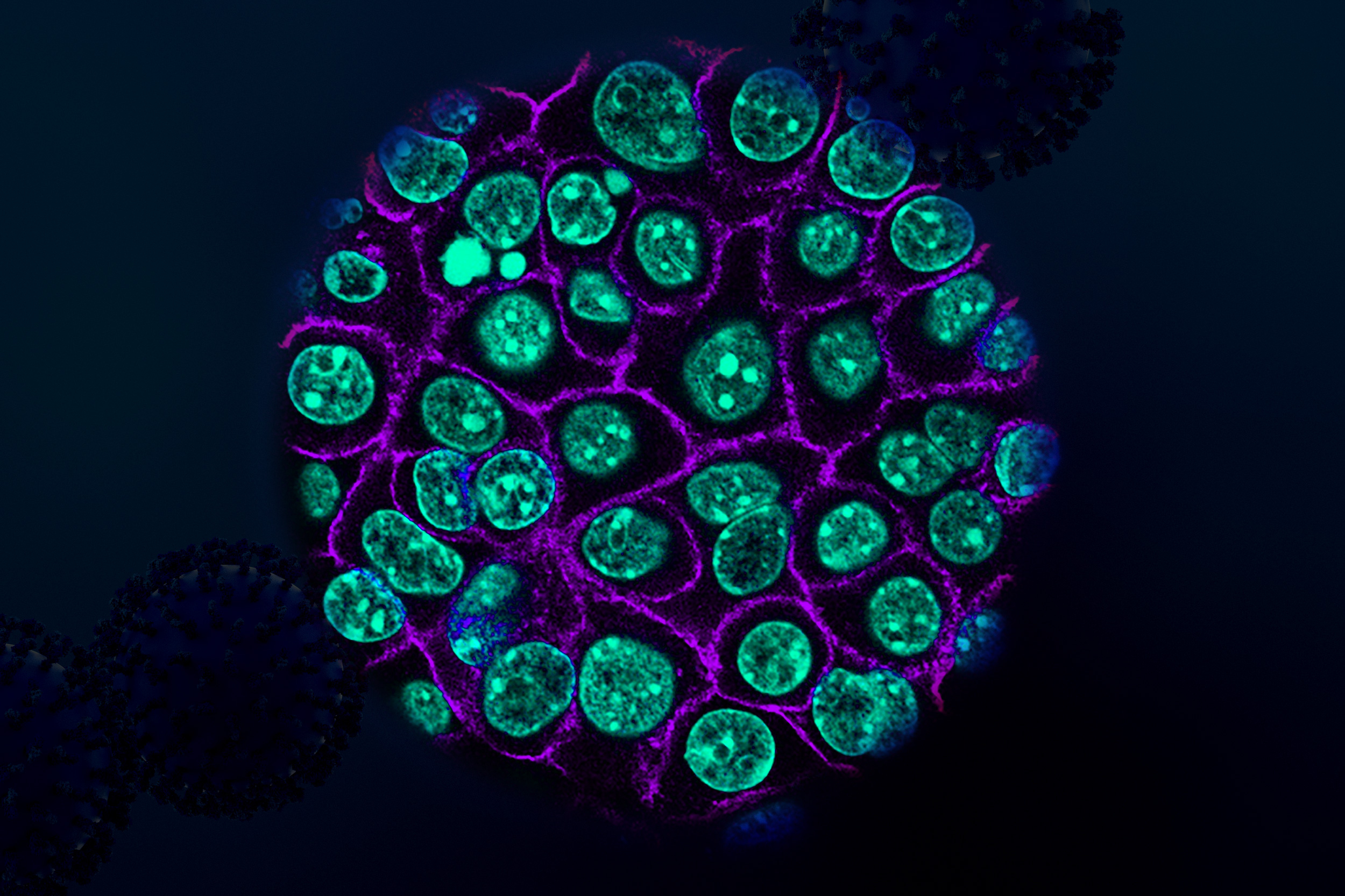
Calo’s work on ribosome formation also led him to explore another cell organelle known as the nucleolus, whose role is to help build ribosomes. In 2023, he found that a gene called TCOF1, which can lead to craniofacial malformations when mutated, is critical for forming the three compartments that make up the nucleolus.
That finding, he says, could help to explain a major evolutionary shift that occurred around 300 million years ago, when the nucleolus transitioned from two to three compartments. This “tripartite” nucleolus is found in all reptiles, birds, and mammals.
“That was quite surprising,” Calo says. “Studying disease-related genes allowed us to understand a very fundamental biological process of how the nucleolus evolved, which has been a question in the field that nobody could figure out the answer for.”