Activating proteins linked to longevity may help to increase endurance and combat frailty in the elderly.
Anne Trafton | MIT News Office
March 22, 2018
As we get older, our endurance declines, in part because our blood vessels lose some of their capacity to deliver oxygen and nutrients to muscle tissue. An MIT-led research team has now found that it can reverse this age-related endurance loss in mice by treating them with a compound that promotes new blood vessel growth.
The study found that the compound, which re-activates longevity-linked proteins called sirtuins, promotes the growth of blood vessels and muscle, boosting the endurance of elderly mice by up to 80 percent.
If the findings translate to humans, this restoration of muscle mass could help to combat some of the effects of age-related frailty, which often lead to osteoporosis and other debilitating conditions.
“We’ll have to see if this plays out in people, but you may actually be able to rescue muscle mass in an aging population by this kind of intervention,” says Leonard Guarente, the Novartis Professor of Biology at MIT and one of the senior authors of the study. “There’s a lot of crosstalk between muscle and bone, so losing muscle mass ultimately can lead to loss of bone, osteoporosis, and frailty, which is a major problem in aging.”
The first author of the paper, which appears in Cell on March 22, is Abhirup Das, a former postdoc in Guarente’s lab who is now at the University of New South Wales in Australia. Other senior authors of the paper are David Sinclair, a professor at Harvard Medical School and the University of New South Wales, and Zolt Arany, a professor at the University of Pennsylvania.
Race against time
In the early 1990s, Guarente discovered that sirtuins, a class of proteins found in nearly all animals, protect against the effects of aging in yeast. Since then, similar effects have been seen in many other organisms.
In their latest study, Guarente and his colleagues decided to explore the role of sirtuins in endothelial cells, which line the inside of blood vessels. To do that, they deleted the gene for SIRT1, which encodes the major mammalian sirtuin, in endothelial cells of mice. They found that at 6 months of age, these mice had reduced capillary density and could run only half as far as normal 6-month-old mice.
The researchers then decided to see what would happen if they boosted sirtuin levels in normal mice as they aged. They treated the mice with a compound called NMN, which is a precursor to NAD, a coenzyme that activates SIRT1. NAD levels normally drop as animals age, which is believed to be caused by a combination of reduced NAD production and faster NAD degradation.
After 18-month-old mice were treated with NMN for two months, their capillary density was restored to levels typically seen in young mice, and they experienced a 56 to 80 percent improvement in endurance. Beneficial effects were also seen in mice up to 32 months of age (comparable to humans in their 80s).
“In normal aging, the number of blood vessels goes down, so you lose the capacity to deliver nutrients and oxygen to tissues like muscle, and that contributes to decline,” Guarente says. “The effect of the precursors that boost NAD is to counteract the decline that occurs with normal aging, to reactivate SIRT1, and to restore function in endothelial cells to give rise to more blood vessels.”
These effects were enhanced when the researchers treated the mice with both NMN and hydrogen sulfide, another sirtuin activator.
Vittorio Sartorelli, a principal investigator at the National Institute of Allergy and Infectious Diseases who was not involved in the research, described the experiments as “elegant and compelling.” He added that “it will be of interest and of clinical relevance to evaluate the effect of NMN and hydrogen sulfide on the vascularization of other organs such as the heart and brain, which are often damaged by acutely or chronically reduced blood flow.”
Benefits of exercise
The researchers also found that SIRT1 activity in endothelial cells is critical for the beneficial effects of exercise in young mice. In mice, exercise generally stimulates growth of new blood vessels and boosts muscle mass. However, when the researchers knocked out SIRT1 in endothelial cells of 10-month-old mice, then put them on a four-week treadmill running program, they found that the exercise did not produce the same gains seen in normal 10-month-old mice on the same training plan.
If validated in humans, the findings would suggest that boosting sirtuin levels may help older people retain their muscle mass with exercise, Guarente says. Studies in humans have shown that age-related muscle loss can be partially staved off with exercise, especially weight training.
“What this paper would suggest is that you may actually be able to rescue muscle mass in an aging population by this kind of intervention with an NAD precursor,” Guarente says.
In 2014, Guarente started a company called Elysium Health, which sells a dietary supplement containing a different precursor of NAD, known as NR, as well as a compound called pterostilbene, which is an activator of SIRT1.
The research was funded by the Glenn Foundation for Medical Research, the Sinclair Gift Fund, a gift from Edward Schulak, and the National Institutes of Health.
March 19, 2018
Whitehead Institute researchers uncover framework for how stem cells determine where to form replacement structures.
Lisa Girard | Whitehead Institute
March 15, 2018
Researchers at Whitehead Institute have uncovered a framework for regeneration that may explain and predict how stem cells in adult, regenerating tissue determine where to form replacement structures.
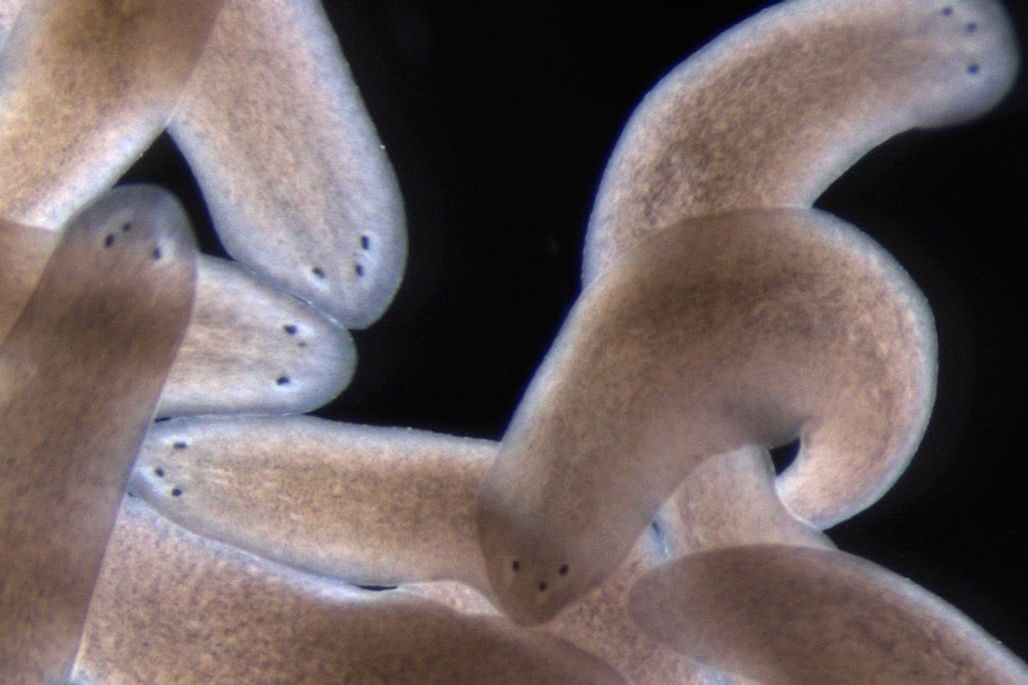
In a paper that appeared online March 15 in the journal Science, the researchers describe a model for planarian (flatworm) eye regeneration that is governed by three principles acting in concert, which inform how progenitor cells behave in regeneration. The model invokes positional cues that create a scalable map; self-organization that attracts progenitors to existing structures; and progenitor cells that originate in a diffuse spatial zone, rather than a precise location, allowing flexibility in their path. These principles appear to dictate how progenitor cells decide where to go during regeneration to recreate form and function, and they bring us closer to a systems-level understanding of the process.
From previous work, the researchers knew that stem cells are likely reading out instructions from neighboring tissues to guide their path, and it became clear that the process faces some serious challenges in regeneration. “We realized that positional information has to move; it needs to change during regeneration in order to specify the new missing parts to be regenerated. This revised information can then guide progenitor cells that are choosing to make new structures to differentiate into the correct anatomy at the correct locations,” says the paper’s senior author Peter Reddien, a Whitehead Institute researcher, an MIT professor of biology, and a Howard Hughes Medical Institute (HHMI) investigator. “There is a puzzle that emerges, however. Since positional information shifts after injury during regeneration, there is a mismatch between the positional information pattern and the remaining anatomy pattern. Realizing this mismatch exists was a trigger for our study. We wanted to understand how stem cells making particular tissues decide where to go and differentiate. Is it based on anatomy, or is it based on positional information? And when those two things are not aligned, how do they decide?”
Reddien and his lab have spent over a decade unraveling the mysteries of regeneration using a small flatworm, called the planarian. If a planarian’s head is amputated, or its side is removed, each piece will regenerate an entire animal. In order to understand how progenitors decide where to go in the noisy environment of animal regeneration, the researchers used the planarian eye, a visible organ that is small enough to be removed without serious injury and has the added advantage of having defined progenitor cell molecular markers.
The researchers devised a simple experiment to resolve the question of how the progenitors decide where to go: amputate the animal’s head, then after three days remove one of the eyes from the head piece. What they found was that progenitor cells would nucleate a new eye in a position anterior (closer to the tip) to the remaining eye, rather than in the “correct” position specified by the anatomy, symmetrical with the current eye. However, if the same experiment is done, except with one of the eyes removed from the head piece earlier — the same day as the amputation, rather than three days later — there is a different result: The new eye is nucleated at a position symmetrical to the remaining eye, at the “correct” position according to the anatomy, suggesting that when the choices are conflicting, anatomical self-organizing dynamics win.
These simple rules guide the system to successful regeneration and also yield surprising outcomes when the system is pushed to its limits, producing alternative stable anatomical states with three-, four-, and five-eyed animals. Resecting both the side of an animal and amputating the head can place progenitors far enough from existing, self-organizing attractors that they miss them, allowing them to nucleate a new attractor — a third eye — in the head fragments. “If the migrating progenitors are too close to the attractor that is the existing eye, it will suck them in, just like a black hole; if they are far enough, they can escape,” says Kutay Deniz Atabay, first author on the paper and a graduate student in the Reddien lab.
When the researchers did the same surgery on a three-eyed animal, the progenitors miss the attraction of existing eyes and form a five-eyed animal. In each of these cases, all of the eyes are functionally integrated into the brain and exhibit the same light-avoidance response of the planarian eye. The researchers also observed that these alternative anatomical states endure, revealing rules for maintenance of existing and alternative anatomical structures. “This recipe helps provide an explanation for a fundamental problem of animal regeneration, which is how migratory progenitors make choices that lead the system to regenerate and maintain organs,” Atabay says.
“These studies contribute to a proposed framework for regeneration in which competing forces, self-organization, and extrinsic cues, are the guideposts impacting the choice of progenitor targeting in regeneration; and those two forces together determine the outcome,” Reddien says.
This work was supported by the National Institutes of Health (NIH) and the HHMI. Additional funding was provided by the MIT Presidential Fellowship Program and a National Defense Science and Engineering Graduate Fellowship.
Clare Harding's microscope image of Toxoplasma gondii parasites is one of this year's winners at the Koch Institute Image Awards
Nicole Giese Rura
March 9, 2018
Scientists use a variety of approaches to unravel the functions of organisms, cells, and even molecules, and some of these approaches produce images that are as stunning as they are informative. Since 2011, the annual Koch Institute Image Awards, conducted by The Koch Institute for Integrative Cancer Research at MIT, has honored outstanding images created by life science and biomedical researchers in the MIT community.
This year, one of the winning pictures was created by Clare Harding, a postdoctoral researcher in the lab of Whitehead Institute Member and MIT assistant professor of biology, Sebastian Lourido. Harding and the other winners were lauded last night at a gala opening of the exhibit on the Koch building’s ground floor where the winning images will be on display for the coming year.
In Harding’s striking entry this year, each white and blue “petal” of the rosettes is a single-celled Toxoplasma gondii, the parasite that causes toxoplasmosis infection. This image was taken moments before the individual parasites comprising the daisy-like clusters would have triggered a massive, coordinated “egress”, which would destroy the host cell they had called home. The host’s nucleus is the large blue oblong jutting in from the upper left (host and parasite DNA are marked blue), and the red dapple marks a molecule in the host cell’s internal skeleton, called tubulin.
Toxoplasmosis infects about 25% of the world’s population and can cause serious disease in pregnant women, infants, and immunocompromised people. Not only is the Lourido lab’s work on T. gondii revealing important clues about this disease, but their research can also shed light on T. gondii’s close cousins on the evolutionary tree: Plasmodium spp., which cause malaria and contribute to more than a million deaths each year; and Cryptosporidium spp., which cause cryptosporidiosis, a gastrointestinal illness that can be fatal in those with a compromised immune system.
Harding’s research in the Lourido lab is focused on GAPM1a, a structural protein that forms a layer directly under T. gondii’s outer membrane and plays a similar architectural role in Plasmodium. This protein scaffold (marked as white in the image) is so vital that it is one of the first things established within daughter cells, which appear in the image as two small white spheres within some of the larger parasites. Parasites lacking the GAPM1a scaffold degrade into amorphous blobs that are unable to infect new host cells—a visual testament of how important this protein is to the parasites.
Light microscopy images like Harding’s are created by passing or reflecting light off of a sample and using one or more lenses to magnify the resulting representation. According to Wendy Salmon, the light microscopy specialist at Whitehead’s W.M. Keck Biological Imaging Facility and a two-time Koch Image Awards judge, all microscopy-based images are imperfect representations of the samples that they depict, because light microscopy is limited by the physics of the light shined on the sample and the glass that comprises the lenses. To push beyond the boundaries of physics and reveal the otherwise invisible, Harding employed two techniques: fluorescent markers and structured illumination microscopy.
Using a light microscope alone, the GAPM1a protein is indiscernible within T. gondii parasites. But by genetically modifying the parasite to produce the GAPM1a protein fused to a green fluorescent protein, Harding could shine a particular wavelength of light onto the sample and cause the fluorescent protein to glow, thereby illuminating GAPM1a’s presence.
In addition to being able to identify the protein she is studying in a sample, Harding has the additional challenge that the parasites are so tiny—5 micrometers in length, or about 1/16th the width of a human hair—that they are beyond the resolution of light microscopy. In order to visualize the GAPM1a scaffold, Harding used a technique called structured illumination microscopy, which takes advantage of the properties of light in order to see things half the size of what is visible with a conventional light microscope. In this technique, the microscope casts a grid of light onto the specimen and takes images as the grid rotates. The resulting data from the images are processed using an algorithm that reconstructs the specimen’s appearance, enhancing its resolution.
Harding has been working with T. gondii for more than three years and microscopy has always played a major role in her work, but her appreciation for the science and art of microscopy has recently flourished.
“I like microscopy partly because it’s beautiful and partly because with a lot of other techniques, you need to interpret the data. With microscopy, you know what you’re looking at is right there,” says Harding, who is thrilled to have her work featured in the Koch Institute Public Galleries. “I definitely fell in love with microscopy right away. The first time I did it, I realized how much there is to a cell. Even just staining the DNA in a cell, suddenly you can see stars.”
Whitehead Institute study in yeast illuminates the role of a molecular de-clogger in disease biology.
Nicole Davis | Whitehead Institute
March 8, 2018
Nearly half a billion people worldwide live with type 2 diabetes. Yet despite the disease’s sizeable and increasing impact, its precise causes remain murky. Current scientific thinking points to two key processes: insulin resistance, wherein cells develop ways of tuning out insulin’s signals, and the breakdown of beta cells, the specialized cells in the pancreas that produce insulin. The molecular bases for these activities, however, are largely unknown.
Now, a team of researchers based at Whitehead Institute is casting new light on the theory that abnormal protein deposits — similar to ones that emerge in neurodegenerative disorders such as Alzheimer’s disease — accumulate in and around beta cells and derail their normal function. The team’s findings, which appear in today’s advance online edition of the journal Cell, illuminate the function of a key protein, called Ste24, which unclogs the cellular machinery that helps shuttle proteins into compartments within the cell. The researchers believe that this unclogging action could be an important way to minimize or even prevent the protein deposits that damage precious beta cells in type 2 diabetes.
“We’ve created a new platform for identifying potential genetic and pharmaceutical targets that can help neutralize the toxic proteins that build up in patients with the disease,” says lead author Can Kayatekin. “In initial studies with this platform, we unveiled a very interesting target, Ste24, which has opened an important window on the biology of proteotoxicity in type 2 diabetes.”
Alongside the glucose-lowering hormone insulin, beta cells produce another protein, called IAPP (short for human islet amyloid polypeptide). As these two proteins mature inside the cells, they are bundled together and released within the same miniature packets, or vesicles. However, as its name suggests, IAPP is very amyloidogenic — that is, prone to forming large aggregates, which can pile up both within and outside of cells.
“What happens is that as demand for insulin increases, you get more and more IAPP production, and the more you make, the more likely it is to aggregate,” Kayatekin says. “So, the idea is that as you make more IAPP, it starts poisoning the very cells that are producing it.”
To further explore the molecular mechanisms of IAPP production and aggregation, Kayatekin harnessed a powerful paradigm established by his late mentor and supervisor Susan Lindquist, a Whitehead Institute member, MIT professor of biology, and HHMI Investigator who passed away in 2016. Her pioneering approach leverages the baker’s yeast Saccharomyces cerevisiae to create models of toxic proteins in order to probe, perturb, and expose their underlying biology.
Kayatekin and the study’s co-authors generated a yeast model that carries six tandem copies of IAPP. “In most of the neurodegenerative and protein aggregation diseases, the research has trended towards these kinds of smaller oligomers, which seem to be more capable of diffusing in the cell and are therefore likely to be more toxic,” he explains.
With their model of IAPP toxicity in hand, the researchers then turned to genetic techniques to identify yeast proteins that either enhance or ameliorate the effects of IAPP aggregation. Kayatekin and his team identified several intriguing finds, perhaps the most interesting one being a protease called Ste24. According to a 2016 study published in Cell by Maya Schuldiner’s laboratory, Ste24 can cleave proteins that clog translocons — the channels through which secreted proteins, including IAPP, must pass before they can be released. Much like liquid drain cleaners can clear household pipes of hair balls and other muck, Ste24 can remove proteins that get stuck as they venture through the cell’s inner straits. Indeed, Kayatekin finds that overexpressing Ste24 in his yeast model can help rescue some of the effects of IAPP deposits.
Notably, Ste24 is highly conserved through evolution — so much so that the human version, ZMPSTE24, can stand in for its yeast counterpart, the researchers found. This remarkable feature allowed the team to begin functionally dissecting how natural variation in the human protein might impact its unclogging function. By scouring different genetic variants in ZMPSTE24 identified with the help of the AMP T2D-GENES Consortium, they discovered versions whose function was impaired. Initial data suggests that some of these loss-of-function mutants may be more common in type 2 diabetes patients than those without the disease — suggesting that a less-than-robust declogger could possibly contribute to type 2 diabetes progression.
More work is needed to fully decipher the biology of Ste24, IAPP toxicity, and type 2 diabetes. Nevertheless, Kayatekin hopes that his innovative yeast model will prove to be as powerful a tool for illuminating the molecular underpinnings of disease as the ones that preceded it.
Funding for this work was provided by Whitehead Institute, the Picower Institute at MIT, the University of Texas, M.D. Anderson Center, the Howard Hughes Medical Institute, the Glenn Foundation for Medical Research, the Eleanor Schwartz Charitable Foundation, the Edward N. and Della L. Thome Foundation, the JPB Foundation, the Robert A. and Renee E. Belfer Foundation, the National Institutes of Health, the Canadian Institute of Health Research, and the U.S. Department of Defense. The researchers received additional support from the American Italian Cancer Foundation, the American Parkinson’s Disease Foundation, and the Helen Hay Whitney Foundation.
MIT biological engineers discover why a promising drug failed in clinical trials.
Anne Trafton | MIT News Office
March 6, 2018
Pharmaceutical companies once considered a protein called p38 a very attractive target for treating rheumatoid arthritis. Arthritis patients usually have elevated activity of this inflammation-producing protein, and in lab studies p38 inhibitors appeared to soothe inflammation. However, these drugs failed in several clinical trials.
A new study from MIT sheds light on just why these drugs did not work for arthritis. By untangling the complex interactions between different cell pathways involved in inflammation, the researchers discovered that shutting off p38 triggers other inflammatory pathways.
The findings demonstrate the importance of studying a potential drug’s impact on complex cellular systems, says Doug Lauffenburger, head of MIT’s Department of Biological Engineering and the senior author of the study. It’s also important to do these studies under environmental conditions that match those found in diseased tissue, he adds.
“You’ve got to make sure you understand the complexity of the intracellular networks, and beyond that, you need to think about the environment you put the cells in,” Lauffenburger says. “It’s easy to get different results in different contexts, so you need to study them under many different conditions.”
Former MIT postdoc Doug Jones is the lead author of the paper, which appears in the March 6 issue of Science Signaling.
A promising target
Rheumatoid arthritis, which afflicts more than 1 million Americans, is an autoimmune disorder that produces swollen and painful joints, primarily affecting the wrists and hands. This pain results from inflammation in the lining of the joints. Cells called synovial fibroblasts, which typically provide structural support for the joint lining, promote the inflammation and swelling in arthritic conditions.
Several years ago, scientists seeking new treatments for arthritis discovered that synovial fibroblasts from arthritis patients had very high levels of p38, and many pharmaceutical companies began working on p38 inhibitors. “The activity of this pathway was so strong that people tended to think that it was the best one to inhibit,” Lauffenburger says.
Despite their promise, p38 inhibitors failed in phase II clinical trials run by at least eight pharmaceutical companies. One of those companies, Boehringer Ingelheim, asked Lauffenburger to help them figure out why. Lauffenburger’s lab focuses on systems biology, a field that involves measuring the interactions of many cell components and then performing computational modeling of those measurements to predict cell behavior.
The researchers’ analysis revealed that the inflammatory pathway controlled by p38 interacts with several other pathways that can cause inflammation. These pathways, known collectively as stress pathways, produce inflammatory cytokines in response to events such as infection or injury.
The MIT team found that when p38 is extremely elevated, it suppresses the activity of these other inflammatory pathways. Therefore, when it gets turned off, the brake on the other pathways is released. Under these circumstances, inflammation remains high — the difference is that now it is controlled by other stress pathways.
“This is an insightful paper on redundancy in signaling and the need to understand compensatory mechanisms before spending billions on drug development. In that sense, it is a far more important insight than ‘just’ p38 inhibitors, and it makes clear again that animal efficacy models have severe limitations as tools to predict human efficacy,” says David De Graaf, CEO and president of Syntimmune, who was not involved in the research. “This paper outlines one very thoughtful and generic approach to answer complex questions about efficacy in ex vivo human model systems.”
Environment matters
Why was the MIT team able to see this phenomenon when others had not? Lauffenburger says one key is the environment in which the synovial fibroblast cells were studied.
Normally, cells studied in the lab are grown in a culture medium that offers them nutrients and molecules called growth factors, which keep the cells alive and proliferating. However, the MIT team found that under these conditions, a pro-growth pathway called MEK actually keeps p38 levels lower than in cells under stress. Because p38 is not as high, it doesn’t inhibit the other stress pathways as strongly, so when the cells are exposed to p38 inhibitors, the other pathways don’t soar into action and overall inflammation goes down.
“It looks like p38 inhibitors work well, if cells are in these growth factor environments,” Lauffenburger says.
However, the MIT team found that synovial fluid from arthritis patients is not a pro-growth environment but is full of inflammatory cytokines. They then decided to expose synovial fibroblasts taken from patients with arthritis and from healthy individuals to this inflammatory environment. In both healthy and diseased cells, p38 levels skyrocketed, producing more inflammation and shutting off other stress pathways.
One question still to be answered is whether p38 inhibitors could work against other diseases such as cancer, in which the cells targeted would likely be in a pro-growth environment. They are also being considered as potential treatments for other inflammatory diseases such as multiple sclerosis and Alzheimer’s. Lauffenburger says that their success will likely depend on what kind of environment the affected cells are in.
“A p38 inhibitor could work; you just have to know what the context is that the target cells are in. If you have the same kind of inflammatory cytokines there, then you might encounter the same problem” seen in arthritis, he says.
It’s also possible that p38 inhibitors could work against arthritis or other drugs if given along with drugs that shut off other stress pathways, but more research would be needed to investigate that possibility, Lauffenburger says.
The research was funded by the National Institutes of Health, the Army Research Office, and Boehringer Ingelheim Inc. The project was undertaken in collaboration with Professor Peter Sorger at Harvard Medical School; Brian Joughin at MIT and Anne Jenney at Harvard were also significantly involved in the work.
Elizabeth Li ’18 has worked in three cancer-related labs over the past six years, and one day intends to start her own.
Raleigh McElvery
March 6, 2018
Senior Elizabeth Li recreates miniature organs — lungs and intestines — outside the body. She does so in order to study cancer progression in both environments, and over the past six years has worked in three separate cancer-focused labs: two at MIT and another beginning her junior year of high school. One day, she aims to run her own.
“I’ve been into math and science ever since I was little,” she explains, “but in third grade I met a friend who was pretty important to me. She was diagnosed with a very malignant form of brain cancer and ended up dying from it. From that point on — even though I was still very young — I knew I wanted to do cancer research.”
In 9th grade, Li began at the School for Science and Math at Vanderbilt, a joint program between the university and Metro Nashville Public Schools. “I got to skip school once a week to learn research techniques, and had the opportunity to join Andries Zijlstra’s lab my junior year,” she recalls. “I’m actually still part of that group, and I’ve been working on the same project related to cancer metastasis for six years now.”
When it came time to select a university, Li was torn between Vanderbilt — where she was already performing research — and MIT, which she describes as “the place of opportunity.” She was ultimately swayed by MIT’s vast array of research areas, and fully sold after an overnight to preview the undergraduate culture.
Li opted for Course 7 in order to continue doing cancer research, and joined Omer Yilmaz’s lab in 2015 to investigate intestinal tumorigenesis. Here, she spent most of her time doing organoid work, studying the progression of colorectal cancer in miniaturized and simplified versions of the intestine. Li removed individual intestinal stem cells — or sometimes the entire “crypts” in which they reside — and grew them inside a 3D gel. This environment allowed the cells to differentiate and interact as they would in the colon, rather than growing on a flat, plastic dish.
Li and other members of the Yilmaz lab watched these cells multiply, observing their shape and the regeneration process. Li’s method of assessment varied depending on the research question: on some days, she stained the cells for proliferation markers, and on others she exposed them to different metabolites or drugs to see how the cells responded.
“On a typical day, I would come in during the morning between classes, and again in the afternoons and evenings,” she says. “The experiments differed, but we tended to do a lot of genetic manipulation. We’d make plasmids, CRISPR-Cas9 knockouts, or test for gene and protein expression using qPCR and Western blots.”
After two years, Li’s primary mentor finished her postdoctoral training, and Li transitioned to Jackie Lees’ lab at the beginning of her senior year. Li is now working with a fellow undergraduate on an independent project, centered on the enzyme protein arginine methyltransferase 5 (PRMT5).
PRMT5 catalyzes the transfer of methyl groups to the amino acid arginine in certain proteins, modifying their function. The enzyme also plays a key role in regulating gene splicing, the process by which segments of pre-mRNA are removed — changing the genetic code so that multiple genes can be encoded by the same initial transcript.
The Lees lab is interested in PRMT5 because it affects glioblastoma formation, the most common form of adult brain malignancy. As Li explains, when PRMT5 expression increases, so does tumor formation. Since there are still relatively few therapeutic options to treat glioblastomas, she’s hoping to use small molecules to inhibit PRMT5 expression and thus impede tumor initiation and progression.
“We’re considering using nanoparticles to deliver them,” she says, “and in doing so, hoping to gain a better understanding of how PRMT5 inhibition might impact cancer progression and tumorigenesis.”
Li is testing one small molecule PRMT5 inhibitor in lung organoids and several 2D cell lines — determining how sensitive the cells are and if the organoids will form, to gauge whether a tumor would still develop in the presence of the drug. “Depending on when you add the drug, you can test different aspects of tumorigenesis,” she explains.
She’s also split the past four years between the Biology Undergraduate Student Association (of which she was faculty liaison, outreach chair, and then co-president), the MIT Pre-Medical Society,MIT Lion Dance, Asian Dance Team, Wind Ensemble, and Improv-a-Do! She’s also heavily involved in DynaMIT, which organizes an annual, week-long science program for economically disadvantaged middle school students.
“There are a lot of extracurriculars to do,” Li admits. “But it’s pretty easy to get involved in the MIT community and still stay on top of your coursework, if you keep it to four or five classes per semester. It’s worked out for me so far — I’m still alive and happy and have time for eating, sleeping, and friends.”
As Li applies to MD-PhD programs, she hopes one day to practice medicine (perhaps pediatric oncology) while running her own lab.
“My advice for incoming MIT undergrads would be to remember to have fun,” she says. “You only have four years, so take advantage of your time here: hang out with your friends, take the classes you want to take, and do things that you enjoy. Hopefully most of those activities will be one and the same.”
Photo credit: Raleigh McElvery
Written by Merrill Meadow | Video by Ceal Capistrano
February 27, 2018
TabulaSynthase The blog of Whitehead Institute; bringing together ideas and perspectives from the Whitehead community and beyond.
As Rudolf Jaenisch and Rick Young sit side-by-side in conversation, a long reptilian skull is perched behind them. Its intact teeth still menacingly sharp, it was a “salty,” a carnivorous, ferocious, salt water crocodile from the north Australian coast. The skull is both a piece of natural history and the perfect souvenir for two scientists who appear most happy when taking on some new and not-wholly-calculable risk—whether as pioneering researchers or globe-trekking adventurers.
Though born half-a-world apart—Jaenisch in Wölfelsgrund, Germany; Young in Pittsburgh, Pennsylvania—the two have collaborated virtually since each arrived at Whitehead in 1984, leveraging shared passions for discovery research and for exploring the Earth’s least-travelled paths. And they have found a potent-but-unexpected synergy between their scientific ventures in Cambridge and their sometimes-risky expeditions to spots such as Chile’s Patagonia, the Namibian desert, and the Himalayas.
Young has developed ground-breaking technology to map human genome regulatory circuitry and discovered core circuitry of human embryonic stem cells; an educator and bio-tech entrepreneur, as well as bench scientist, he’s been recognized as one of the top 50 leaders in science, technology and business. Jaenisch, a National Medal of Science (NMS) recipient, developed technology to create mice with virtually any genetic mutation an investigator wants to study, and is uncovering “druggable” aspects of the mechanisms underlying infectious, autoimmune, and neurological diseases. Together, the two have pursued the investigations underpinning a dozen of the world’s most cited research papers—including a series of studies that laid the groundwork for understanding the genetic control of stem cell pluripotency.
To paraphrase the NMS award announcement, Jaenisch embodies fearlessness even as he explores the very frontiers of human knowledge. “That’s right on the mark,” Young muses. “The key trait that Rudolf exhibits in both science and travel is that he is fearless.”
“Yes, but I often rely on Rick to get me out of trouble,” Jaenisch jokes, recalling an experience from their trip through the Namibian desert seven years ago. On a whim, they decided to climb a granite mountain that seemed to rise straight out of the desert. Local guides said it was a three-day climb; Jaenisch and Young calculated they could do it in one, if they started well before dawn and finished before dusk (to avoid night-hunting leopards). And so they did, but not without incident. Just after their mid-day break, feeling a concern he couldn’t define, Young insisted that they start back down, even though they were close to the summit. “We argued about it for a while, because I really wanted to reach the top,” Jaenisch says. “But one of our standing rules is that whatever we do, we do together, and Rick was adamant about not going to the top, so we turned around.” Even so, their return proved dicey. They had to minutely ration their water for the last six hours; and Jaenisch was virtually crawling by the time they reached their vehicle. “Rick was probably right,” he says, wryly.
“On our treks, Rudolf has no sense of danger, whereas I’m the guy who gets a little anxious sometimes,” Young says. Those protestations aside, his anxiety sounds like the internal voice of the experienced pilot that he is, used to making instinctual calculations, quickly. And the fact that Young is only somewhat less inclined to risk-taking becomes clear when he discusses his choice on their Australia trip to go for a morning run beside waters harboring fast-running salt crocs.
Helping each other get out of challenging situations—such as when they and fellow climbers were caught in a blinding snow-storm on a Himalayan peak—has created both deep trust and fine-tuned understanding of each other’s capacities. As a result, explains Young, “When we’re back at Whitehead doing science, we have very good instincts about what we can accomplish together and we’re not afraid to do things that are, scientifically, very risky.”
While they make no specific plans to discuss science on their trips—they make as few advance decisions as possible about what they’ll do during a trip—science naturally arises, and some of their most important projects have resulted from these free-wheeling conversations. The most recent include a major research initiative on gene control in diabetes and a proof-of-principle study on a Rett syndrome treatment.
“These trips—whether climbing mountains, kayaking the open ocean, or hiking barely explored forests—are all about four things: exploring places we’ve never seen, learning something new, testing our physical capacities, and wholly unplugging from the rest of the world,” Jaenisch explains.
“We go with our eyes, ears, and minds wide-open. In that context, especially with someone who you really trust, it’s natural that good ideas emerge and that some of them be quite risky. But the science that’s emerged from our excursions has been a wonderful serendipity.”
Matthew Vander Heiden helped revive the forgotten— but critical—study of cancer metabolism.
Sam Apple | MIT Technology Review
February 21, 2018
One day last October, MIT biology professor Matthew Vander Heiden showed up in one of his trademark plaid shirts to teach his undergraduate course on cancer biology. As usual, he peppered his lecture with questions, filling six sliding chalkboards with arrows mapping cellular pathways; he had to erase the boards halfway through class to make room for more notations. But what might have seemed like an ordinary lecture was far from ordinary in one respect: although Vander Heiden was explaining some of the most fundamental aspects of how tumors grow, most of what he was teaching his students would have been absent from nearly every introductory course on cancer biology a decade ago. The science Vander Heiden discussed that afternoon amounted to a once-lost but recently rediscovered chapter in the history of cancer research.
What he didn’t mention in class is that he’d played as large a role as anyone in bringing it back.
That lost chapter focuses on metabolism, and how cancers use nutrients for energy and as building blocks for new cancer cells. It began with a discovery in the early 1920s that most cancers stuff themselves with glucose and then use it in an unusual way. Whereas normal cells typically break down glucose by burning it with oxygen, cancer cells extract much of its energy through fermentation—essentially the same process microorganisms use to make yogurt, beer, and other foods. Indeed, early-20th-century researchers noticed that cancer cells seemed to behave more like yeast than the cells of an animal. But though it would briefly become a major school of cancer research, metabolism fell by the wayside in the 1960s as researchers turned their attention to how cancer-causing genes signal cells to divide.
Cancer metabolism research appeared to be dead, until Vander Heiden helped launch its revival around two decades ago. Today it’s among the hottest areas of the field, spawning conferences, journals, and promising new therapies. And it has fundamentally changed the way many researchers understand cancer and its origins.
Modest revolutionary
Metabolism’s downfall as a research area in the late 20th century was largely a reflection of the faddish nature of science. It didn’t help that Otto Warburg, the German scientist who discovered the unusual metabolism of cancer cells, was so arrogant that much of the scientific community disliked him. So it’s probably a good thing that Vander Heiden, a down-to-earth type who’s been known to downplay his own role on research papers to give his students and postdocs first-author billing, has been so central to the metabolism revival.
Vander Heiden, 45, grew up in Port Washington, Wisconsin, a small town on Lake Michigan once known for its lawnmower factories, and he lives up to every stereotype of his background. “He carries his Midwestern sensibilities with him everywhere he goes,” says his wife, Brooke Bevis, a biologist and the operations manager for Vander Heiden’s lab at MIT’s Koch Institute for Integrative Cancer Research. “I finally made him give up my old 1995 Honda Civic just a few years ago.”
When Vander Heiden enrolled at the University of Chicago in 1990, medicine was already on his mind. His younger brother had suffered from a rare blood disorder as a child, and Vander Heiden spent much of his own childhood hanging around children’s hospitals. But he had little thought of becoming an academic scientist until he began a work-study job washing out equipment in a University of Chicago biology lab. The work was not glamorous but came with a perk: the graduate students in the lab would let Vander Heiden make solutions for them and show him how they did their experiments.
After graduating, he enrolled in Chicago’s MD-PhD program and landed in the lab of Craig B. Thompson. Today Thompson is the president and CEO of the Memorial Sloan Kettering Cancer Center, but at the time he was studying immunology, looking at how the body eliminated huge numbers of immune cells once they were no longer needed.
When Vander Heiden arrived in Thompson’s lab in 1996, part of the explanation was already understood. Those cells would simply commit suicide, a process known as apoptosis. It was also known that a family of proteins named Bcl-2 could stop a cell from committing suicide—and that they appeared to do so through their impact on mitochondria, tiny organelles known as the powerhouses of the cell for their role in energy production.
Vander Heiden had just joined a cutting–edge immunology lab interested in protein signaling. Yet he had been asked to investigate how Bcl-2 proteins affect mitochondria, a relic of the old, outdated metabolism research. When it became clear that no one in the lab knew much about metabolism, Vander Heiden reread the relevant sections of his undergraduate biochemistry textbook. He also teamed up with Navdeep Chandel, a metabolism researcher at Northwestern University who was then a graduate student in a University of Chicago cellular physiology lab.
When another lab showed that proteins released from the mitochondria could trigger apoptosis, Vander Heiden and -Chandel got an important clue: the decision to commit suicide could now be traced directly to the mitochondria. And yet the deeper question of what was happening inside them remained mysterious until the two researchers arrived at an answer, thanks to a series of elegant experiments designed by Vander Heiden (whom Chandel calls “a world-class experimentalist”) to study how molecules moved through the mitochondrial membrane. They discovered that the release of the mitochondrial proteins was a sign of a failing powerhouse, a notice to the cell that a brownout was under way so it was time to abort. But brownouts weren’t inevitable; the Bcl-2 proteins, like emergency workers called to the scene of an imminent disaster, could resuscitate the metabolic function of the mitochondria and keep things from getting to that point. The suicide signal, in turn, would never be released.
Daniel Schmidt, a postdoc in Vander Heiden’s lab, prepares cells to study how metabolism affects cancer cell proliferation. Credit: BUCK SQUIBBDaniel Schmidt, a postdoc in Vander Heiden’s lab, prepares cells to study how metabolism affects cancer cell proliferation.
BUCK SQUIBB
For Vander Heiden, this was a “watershed moment.” Among other things, it meant that metabolic enzymes weren’t merely supplying energy from food. Metabolism was governing the most fundamental decision a cell has to make—whether to live or die. That meant it had to be interwoven into the signaling cascades that molecular biologists studied. His feeling at the time, he recalls, was “Oh my goodness. We don’t really understand metabolism.”
Vander Heiden might not have envisioned himself delving into research areas that had been discarded decades earlier, but what was more surprising was how little research was then being done in an area that was “as fundamental as you get in terms of how biology works,” he says. “I looked around and no one was studying it.”
Thompson, recognizing the opportunity, shifted the focus of his lab to metabolism. Vander Heiden, meanwhile, continued to pursue Thompson’s broader question of how the body eliminates unwanted immune cells. He already knew that growth factors, messages sent from one cell to the next, kept cells from committing suicide, but how the signals delivered their survival message remained unclear. What he discovered in a series of studies carried out in the late ’90s followed perfectly from his previous research. Growth factors kept cells alive by giving them permission to eat. Without that permission, a cell soon faced an energy crisis, and the mitochondria released their death signals.
The takeaway was clear: our bodies eliminate unwanted cells by starving them to death.
Solving the metabolism mystery
As Vander Heiden’s MD-PhD program was coming to an end, he hadn’t yet begun to focus on cancer, but its possible links with his research on cell suicide were intriguing. Cancer cells were the other side of the coin—cells that were resistant to suicide, that no longer cared about instructions from other cells. So in 2004, after completing a residency in oncology at Brigham and Women’s Hospital in Boston, he was anxious to investigate cancer metabolism for his postdoc research.
Finding the right lab wasn’t easy. At the time, telling leading researchers he wanted to study how cancer cells consumed glucose was like approaching a high-tech manufacturer and announcing you wanted to study the trucks that brought fuel to the factory. It sounded, Vander Heiden says, “like a really ridiculous thing.”
Vander Heiden eventually found a home in the Harvard lab of Lewis Cantley, who now directs the Meyer Cancer Center at Weil Cornell. His research in Cantley’s lab would help solve one of the central riddles of cancer metabolism: why cancer cells are so ravenous for glucose. Researchers had once assumed that cancer cells were turning to fermentation because they’d lost the ability to use oxygen properly and needed some other way to produce energy. But Vander Heiden’s research on a mutated form of the enzyme pyruvate kinase showed something else. Rather than being used for energy, much of the glucose was being shunted into pathways used to build new molecules. What a growing cancer needs most of all from its food, the research suggested, is more spare parts—raw materials for making new DNA, membranes, and proteins.
Rethinking chemotherapy
Vander Heiden’s research with Cantley would also lead to his involvement with Agios Pharmaceuticals, the company behind one of the most promising new drugs to emerge from the metabolism revival. (Cantley says he played a major role in building the company’s science in its early days.) The drug, AG-221, treats acute myelogenous leukemia, a cancer of the blood and bone marrow. It works by blocking the product of a mutated form of the mitochondrial enzyme IDH-2. Approved by the US Food and Drug Administration in August, it has been hailed as the first real advance for the disease in 30 years.
The approval of AG-221 isn’t the only thing generating excitement in the cancer world. Unlike almost all other cancer drugs, AG-221 doesn’t kill the cancer cells but, rather, allows them to develop out of their deranged state into noncancerous, mature, functioning cells. That a single metabolic enzyme could have such profound effects on which genes are expressed in a cell is now one of the many signs that changes in metabolism are not just a response to the needs of a growing cancer. Often, they may actually be causing the cancer itself. It represents a major shift in thought: many cancer-causing genes long known for their ability to signal cells to keep dividing have now been shown to have additional roles in signaling cells to keep eating. Some researchers now believe the overeating typically comes first, driving the transformations that follow.
Since his arrival at MIT and the opening of his lab at the Koch Institute in 2009, Vander Heiden has treated cancer patients and continued to search for better therapies. In recent years he has focused on improving understanding of chemotherapy. Though typically thought of as general poisons, most chemotherapy drugs work because they disrupt metabolic functions. That much has long been known, but less clear is why a particular drug works for some cancers and not for others, even when two cancers carry the same mutations.
It was while explaining to his undergrad cancer biology students how targeted drugs work that Vander Heiden first thought of an answer. As a cancer doctor, he knew that chemotherapies are often chosen on the basis of where in the body a tumor first arose, but what was it about this location that made the difference?
Vander Heiden’s research in mice now suggests that the answer may lie in which foods are available to the cancer as it forms. Melanoma and colon cancer, for example, often have the same mutations, and yet, as he explains, because the two cancers “grow in very different places in the body,” they likely “have access to different nutrients.” He adds, “It has nothing to do with the genetics.” If he turns out to be right, it could lead to a fundamental change in how oncologists think about which drugs to give their patients.
As Vander Heiden turns his attention to old chemotherapy drugs, rethinking why and how they work, he is once again looking to the past for new insights on cancer. It might be more than a coincidence. As Bevis, his wife, says, the outdated Honda Civic isn’t the only item he has struggled to let go of. “The list goes on and on,” she says. “He hates waste and will use items long after someone else would have replaced them with a newer, shinier model.”
New finding suggests differences in how humans and bacteria control production of DNA’s building blocks.
Anne Trafton | MIT News Office
February 20, 2018
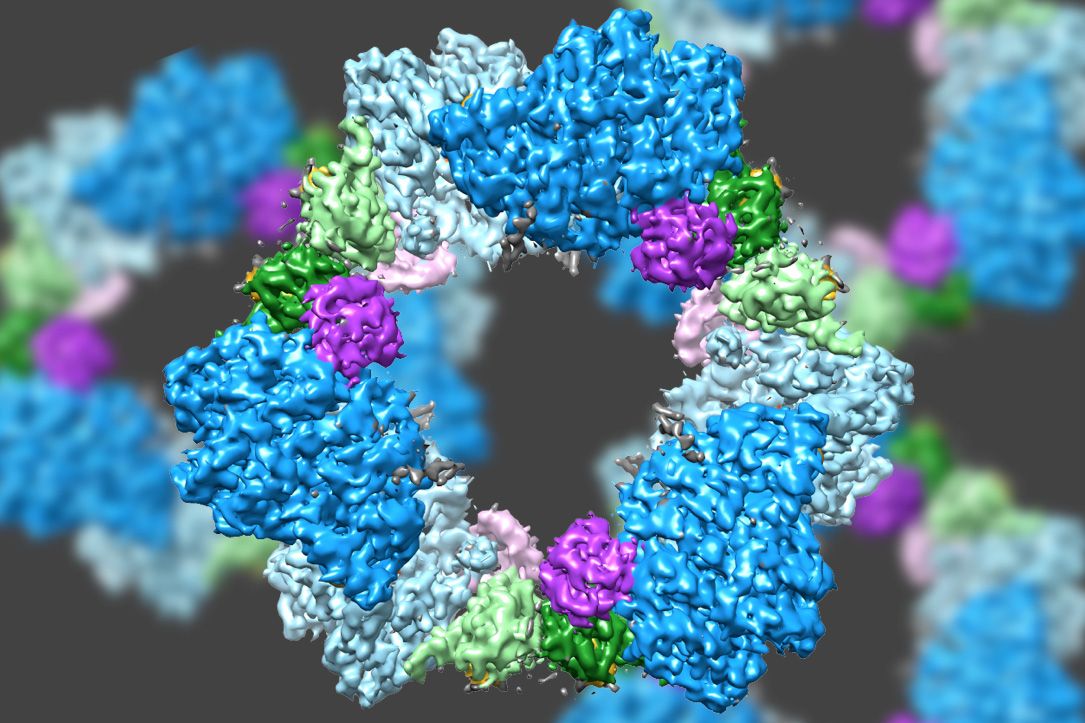
Using a state-of-the-art type of electron microscopy, an MIT-led team has discovered the structure of an enzyme that is crucial for maintaining an adequate supply of DNA building blocks in human cells.
Their new structure also reveals the likely mechanism for how cells regulate the enzyme, known as ribonucleotide reductase (RNR). Significantly, the mechanism appears to differ from that of the bacterial version of the enzyme, suggesting that it could be possible to design antibiotics that selectively block the bacterial enzyme.
“People have been trying to figure out whether there is something different enough that you could inhibit bacterial enzymes and not the human version,” says Catherine Drennan, an MIT professor of chemistry and biology and a Howard Hughes Medical Institute Investigator. “By considering these key enzymes and figuring out what are the differences and similarities, we can see if there’s anything in the bacterial enzyme that could be targeted with small-molecule drugs.”
Drennan is one of the senior authors of the study, which appears in the Feb. 20 issue of the journal eLife. JoAnne Stubbe, the Novartis Professor of Chemistry Emerita at MIT, and Francisco Asturias, an associate professor of biochemistry at the University of Colorado School of Medicine, are also senior authors. The paper’s lead authors are MIT research scientist Edward Brignole and former Scripps Research Institute postdoc Kuang-Lei Tsai, who is now an assistant professor at the University of Texas Houston Medical Center.
An unusual enzyme
The RNR enzyme, which is found in all living cells, converts ribonucleotides (the building blocks of RNA) to deoxyribonucleotides (the building blocks of DNA). Cells must keep a sufficient stockpile of these building blocks, but when they accumulate too many, RNR is shut off by a deoxynucleotide molecule known as dATP. When more deoxynucleotides are needed, a related molecule called ATP binds to RNR and turns it back on.
An unusual feature of RNR is that it can catalyze the production of four different products: the nucleotide bases often abbreviated as A, G, C, and T. In 2016, Drennan discovered that the enzyme achieves this by changing its shape in response to regulatory molecules.
Most of the researchers’ previous work on RNR structure has focused on the version found in E. coli. For those studies, they used X-ray crystallography, a technique that can reveal the atomic and molecular structure of a protein after it has been crystallized.
In the new study, Drennan and her colleagues set out to examine the human version of RNR. This protein’s structure, which turned out to be very different from the bacterial version, proved elusive using X-ray crystallography, which doesn’t work well for proteins that don’t readily crystallize. Instead, the researchers turned to an advanced form of microscopy known as cryo-electron microscopy (cryo-EM).
Until recently, cryo-EM typically offered resolution of about 10 to 20 angstroms, which might reveal the overall shape of a protein but no detail about the position and shape of smaller structural units within it. However, in the past few years, technological advances have led to an explosion in the number of structures achieving resolutions of about 3 angstroms. That is high enough to trace individual protein chains within the larger molecule, as well as internal structures such as helices and even side chains of amino acids.
Scientists already knew that RNR consists of two protein subunits known as alpha and beta. Using cryo-EM, the MIT team found that the human version of the enzyme forms a ring made from six of the alpha subunits. When ATP, which activates RNR, is bound to the enzyme, the ring is unstable and can be easily opened up, allowing the beta subunit to make its way into the ring. This joining of alpha and beta allows the enzyme’s active site, located in the beta subunit, to perform the chemical reactions necessary to produce deoxynucleotides.
However, when the inhibitor dATP is present, the ring becomes much more rigid and does not allow the beta subunit to enter. This prevents the enzyme from catalyzing the production of deoxynucleotides.
Designing drugs
Several cancer drugs now in use or in development target the human version of RNR, interfering with cancer cells’ ability to reproduce by limiting their supply of DNA building blocks. The MIT team has found evidence that at least one of these drugs, clofarabine diphosphate, works by inducing the formation of rigid 6-unit alpha rings.
This 6-unit ring is not found in the bacterial form of RNR, which instead assembles into a distinct ring containing four alpha subunits and four beta subunits. This means it could be possible to design antibiotics that target the bacterial version but not the human version, Drennan says.
She now plans to investigate the structures of other protein molecules that are difficult to study with X-ray crystallography, including proteins with iron sulfur clusters, which are found in many metabolic pathways. The microscopy work in this study was performed at the Scripps Research Institute, but when MIT’s new MIT.nano building opens, it will house two cryo-EM microscopes that will be available to the MIT community as well as other potential users in industry and academia.
“The technological advances that have allowed cryo-EM to get to such high resolution are really exciting,” Drennan says. “It’s really starting to revolutionize the study of biology.”
The research was funded by the National Institutes of Health.





 One day last October, MIT biology professor Matthew Vander Heiden showed up in one of his trademark plaid shirts to teach his undergraduate course on cancer biology. As usual, he peppered his lecture with questions, filling six sliding chalkboards with arrows mapping cellular pathways; he had to erase the boards halfway through class to make room for more notations. But what might have seemed like an ordinary lecture was far from ordinary in one respect: although Vander Heiden was explaining some of the most fundamental aspects of how tumors grow, most of what he was teaching his students would have been absent from nearly every introductory course on cancer biology a decade ago. The science Vander Heiden discussed that afternoon amounted to a once-lost but recently rediscovered chapter in the history of cancer research.
One day last October, MIT biology professor Matthew Vander Heiden showed up in one of his trademark plaid shirts to teach his undergraduate course on cancer biology. As usual, he peppered his lecture with questions, filling six sliding chalkboards with arrows mapping cellular pathways; he had to erase the boards halfway through class to make room for more notations. But what might have seemed like an ordinary lecture was far from ordinary in one respect: although Vander Heiden was explaining some of the most fundamental aspects of how tumors grow, most of what he was teaching his students would have been absent from nearly every introductory course on cancer biology a decade ago. The science Vander Heiden discussed that afternoon amounted to a once-lost but recently rediscovered chapter in the history of cancer research.