Many of the basic biological processes that allow seeds of global food staples like wheat, rice, and corn, to grow, transport nutrients, and develop useful traits like withstanding heat and drought are not yet fully understood. A new gene expression map of seed development offers a framework to better understand, and even guide, seed development to improve crop productivity.
Shafaq Zia | Whitehead Institute
May 19, 2026
Seeds like wheat, rice, and corn are at the center of the global food supply and provide most of the daily calories consumed worldwide. But despite their importance, scientists still do not fully understand many of the basic biological processes that allow these seeds to grow, transport nutrients, and develop traits that determine crop resiliency.
With fluctuating environmental conditions and other stressors threatening agriculture, there is a need to develop hardier crops better able to withstand heat, drought, and changing soil conditions. Scientists are increasingly looking to understand the hidden biology of seed development that could one day help them achieve this.
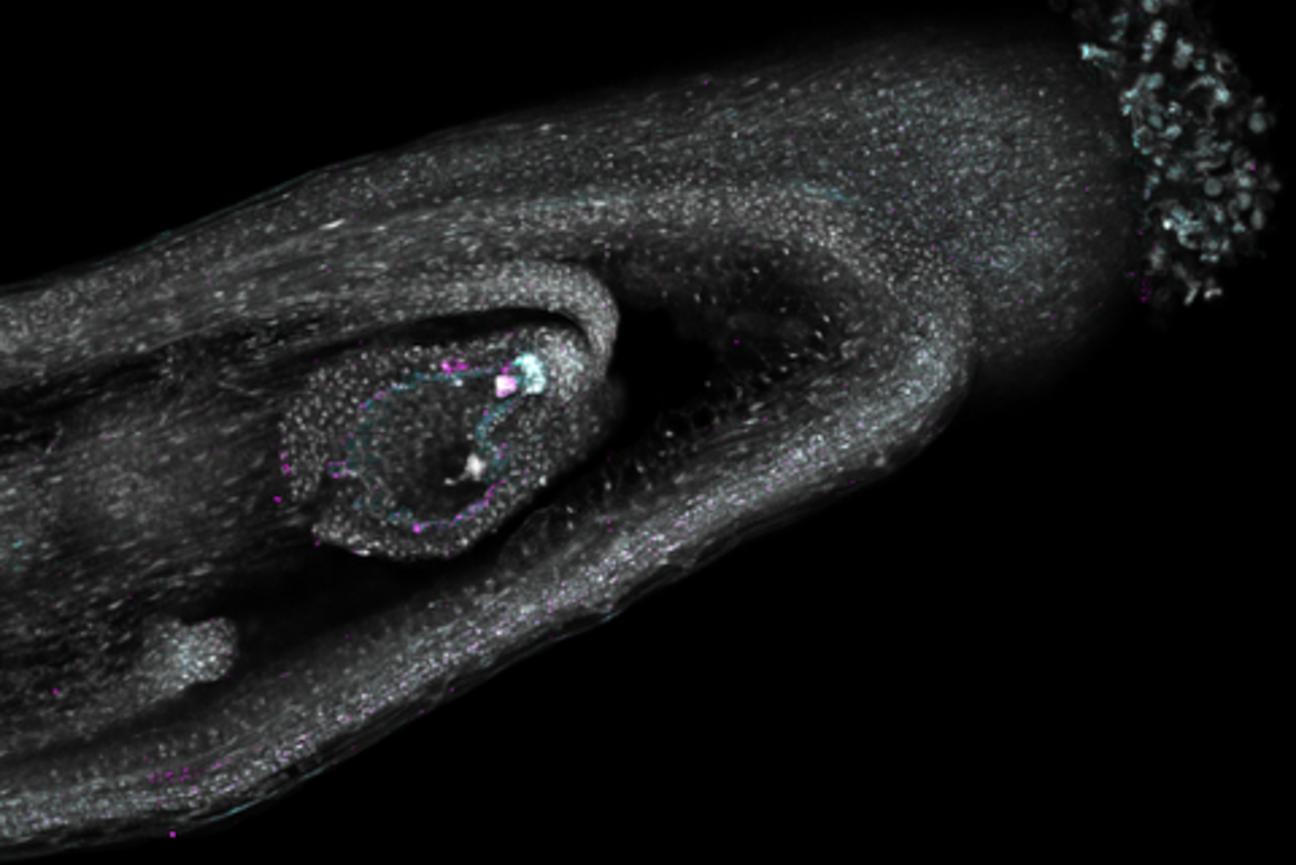
Now, researchers in the lab of Mary Gehring have created a detailed gene expression “map” of seed development in Arabidopsis thaliana, a small flowering plant in the mustard family that is widely used to study plant biology and is closely related to major crops like canola.
This map, also known as a transcriptional atlas, shows which genes are turned on or off in different cell types as the seed develops. Active genes make messenger RNA (mRNA) that guides the production of proteins necessary for cellular processes. By tracking which genes are active where, researchers can better understand the role each cell type plays across different stages of seed development.
The work, published May 21 in Nature Plants, offers scientists new clues about how plants coordinate key biological processes tied to agriculturally significant traits, including seed size and nutrient storage.
“Seeds are fundamental to sustaining human life,” says Caroline (Carly) Martin, lead author of the paper and a graduate student in the Gehring Lab. “By building this atlas, we now have a framework researchers can use to start asking much more precise questions about how seeds develop and if those processes might eventually be improved in different crops.”
Unlike previous atlases of Arabidopsis, which do not distinguish many cell types due to technological limitations, the new atlas provides a more complete and higher resolution view of the developing seed. The researchers have captured seed development at three precisely timed stages after pollination when the plant embryo, the nutrient-rich tissue that feeds it (called the endosperm), and the surrounding tissues from the mother plant rapidly grow and reorganize. Using this dataset, they have identified where genes that regulate how seeds grow and store nutrients are active.
The researchers have found a small group of cells near the plant embryo that activate genes involved in producing brassinosteroids, plant hormones that regulate growth. Previous studies had shown that disrupting the production of this hormone can reduce seed size, but it was not known where within the developing seed the hormone is made.
The new data shows that these hormone-producing cells sit directly next to cells in the endosperm that might respond to the hormone. This close arrangement suggests the two cell types may work together to help fine-tune seed size.
The atlas has also revealed that the endosperm, which nourishes the embryo during development and later becomes the edible portion of many staple crops, contains far more specialized cell types than previously understood by researchers.
The team has identified a small “founder” population of cells that may help establish a key region of the endosperm located at the boundary where nutrients enter the seed from the mother plant.
Because the amount and timing of resources supplied by the mother plant determine how much energy the seed can store, this region of the endosperm helps shape the seed’s nutritional profile. These reserves — oils, starches, and proteins — are essential for both seed development and human nutrition.
These findings, taken together, could allow researchers to better understand — and even guide — seed development to improve crop productivity.
“We’re already seeing that seed filling in many crops is vulnerable to heat stress,” says Gehring, who is also a professor of biology at MIT and an investigator at the Howard Hughes Medical Institute (HHMI). “If we are to solve the humanitarian crises of food insecurity and malnutrition, we need to understand, at a fundamental level, how seeds of different crops form, store nutrients, and survive environmental stress.”
Caroline A. Martin, Kylee R. Cogdill, Alesandra L. Pusey, and Mary Gehring. “A transcriptional atlas of early Arabidopsis seed development suggests mechanisms for inter-tissue coordination.” Nature Plants, May 21, 2026. https://doi.org/10.1038/s41477-026-02295-8