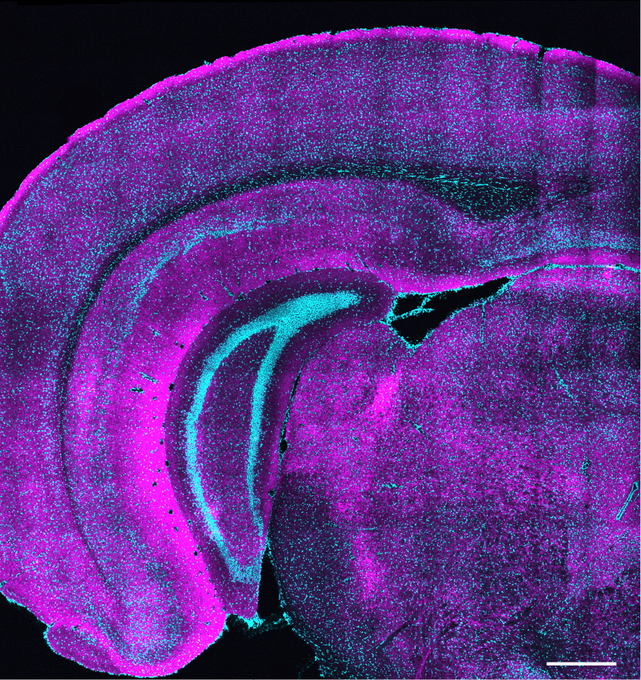
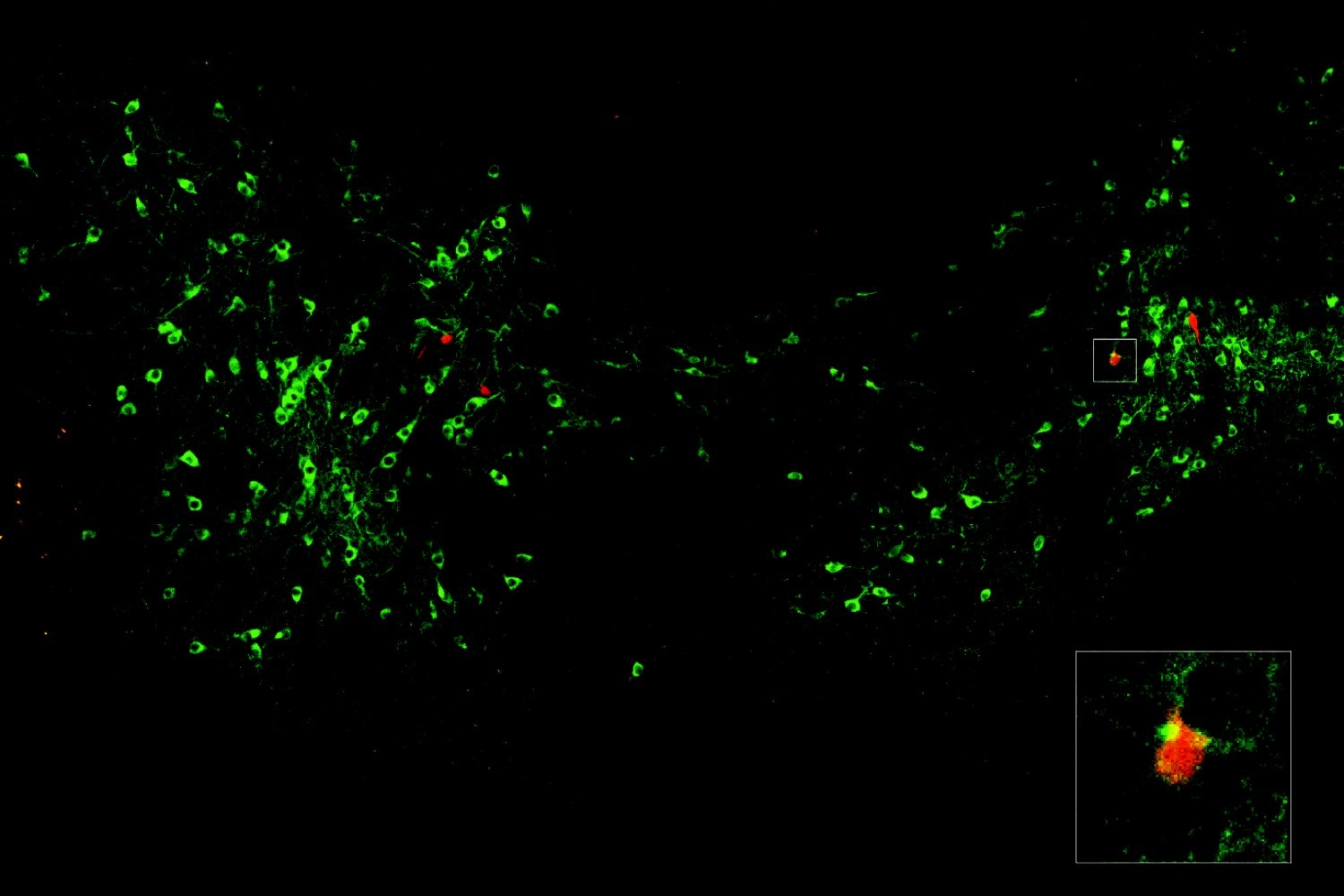
Zuri Sullivan studies sickness behavior to understand how the immune system communicates with the brain to produce changes during illness, hoping to learn more about how the brain interprets immune signals, how these responses may help organisms fight infection, and what they could reveal about disease and immunity.
Shafaq Zia | Whitehead Institute
April 30, 2026
Now, in a new perspective published in Trends in Immunology on April 30, Whitehead Institute Member Zuri Sullivan and colleagues propose a different way of thinking: what if these behaviors are part of an integrated immune strategy that operates across scales — from individual cells to tissues and organs, to the whole organism — and helps promote survival?
Sullivan studies “sickness behavior” to understand how the immune system communicates with the brain to produce these changes during illness — and what they can reveal about how the body coordinates its defense. This work points to a broader biological question: how living systems, from single cells to whole organisms, detect and respond to threats.
We sat down with Sullivan to learn more about how the brain interprets immune signals, how these responses may help organisms fight infection, and what they could reveal about disease and immunity. This interview has been edited for length and clarity.
Whitehead Institute: What led you to start thinking about sickness behavior as a form of whole-organism immunity?
Zuri Sullivan: In graduate school, I found that immune cells in the intestine do more than defend against pathogens — they also help regulate how the body responds to food by changing how intestinal tissue functions depending on the diet.
That work shifted how I thought about immunity, from a local defense system to something broader: a whole-body program that helps shape how we interact with the environment in ways that support survival, including avoiding foods that are harmful or allergenic.
That idea stayed with me in my postdoctoral work in neuroscience, where I studied sickness behavior — things like reduced appetite and social withdrawal during infection. I was interested in how inflammation affects behavior, especially through the hypothalamus, a brain region that controls many of the body’s responses during illness.
Putting those two lines of work together — immunology and neuroscience — led me to an integrated view in which immunity operates across scales, shaping both bodily function and behavior as part of a coordinated system.
WI: We often think of the brain and immune system as separate systems. How are they connected, and why does this connection matter?
ZS: For a long time, the brain was thought to be mostly separate from the immune system, protected by what’s called the blood–brain barrier, which tightly controls what can enter the brain from the bloodstream. That barrier is still very important, but we now know the brain isn’t isolated. The brain and immune system communicate with each other, and that communication can influence both brain activity and behavior. This connection is called the brain–immune axis.
The brain–immune axis is one of the ways the body senses and responds to what’s happening in the outside world. The nervous system does this through our senses, while the immune system uses molecular sensors to detect pathogens and other signs of danger.
The two-way communication between these systems helps coordinate how the body responds to threats. We see this most clearly during infection, in what’s called sickness behavior — things like loss of appetite, fatigue, or social withdrawal. But this connection also matters beyond infection, including in conditions like long COVID and the effects of chronic inflammation on the brain.
In our work, we try to construct a bigger picture of how the body protects itself. Individual cells can defend themselves, tissues like the gut can mount local immune responses, and the brain–immune axis represents the highest level of this system, where the immune system and the brain coordinate to affect both physiology and behavior across the whole body as part of a unified defense response.
WI: Is the brain–immune axis disrupted in chronic diseases like long COVID or other neuropsychiatric disorders?
ZS: In some conditions, the immune response that is normally helpful can become dysregulated. This can happen after infections or due to genetic and environmental factors. When that happens, it can lead to chronic inflammation that starts to damage tissues—for example, scarring in the lungs after infection, or conditions in the gut like inflammatory bowel disease (IBD) or irritable bowel syndrome (IBS).
There are still two main possibilities being studied for long COVID. One is that a small amount of virus remains in the body and keeps the immune system activated. The other is that the virus is gone, but the brain–immune axis becomes dysregulated and keeps the immune system in an activated state. Researchers are still working to distinguish between these two.
What’s also striking is that there are strong associations between inflammation and both neurodevelopmental and neuropsychiatric disorders. For example, people with autism have higher rates of inflammatory gut conditions like IBD and IBS, and many also experience gastrointestinal symptoms. People with IBD and IBS are associated with being at a higher risk of developing anxiety and depression, especially during a flare-up.
What this suggests is that brain–immune communication can influence both brain function and body function in both directions. The challenge now is figuring out causality — whether inflammation drives changes in the brain, the brain drives inflammation, or if it’s a feedback loop between the two.
WI: How can your proposed framework inform how we think about treating infections in the clinic?
ZS: I think it can inform treatment in a few ways. Right now, when people get sick, we often focus on treating symptoms: reducing fever with medications like Tylenol, overriding behaviors like reduced appetite by providing nutrition through feeding tubes in critically-ill patients. But if sickness behavior is part of an organized response, then it becomes important to understand what these behaviors are actually doing before deciding when to suppress them and when to support them.
A useful example comes from a 2016 mouse study. Researchers found that force-feeding sick mice using feeding tubes had a different outcome based on the type of infection they had. Mice with a bacterial infection became more likely to die, but mice with a viral infection had improved survival. What this tells us is that behavioral changes like reduced appetite may actually be tuned to the type of immune challenge the body is facing. So, if we could understand how these behavioral changes affect the course of infection, it could help clarify which interventions are helpful and which might interfere with recovery.
There are also implications beyond acute infection, especially for conditions like long COVID and other neuropsychiatric or post-inflammatory disorders. One key possibility is that the immune system is playing a causal role in either triggering or maintaining some of these conditions. If that’s the case, it becomes especially relevant that the immune system is highly “druggable”— there are already many therapies that target immune pathways. So, understanding how immune signals influence the brain could open up new ways to intervene in conditions where current treatments aren’t working for patients.
What we need is a better map of how different infections affect the brain over time—what we might call “neural signatures” of infection. In animal studies, where we can track both immune responses and brain activity over time, we can start to build that kind of map: how you go from a healthy state and through infection to changes in brain function and behavior.
The hope is that this kind of framework would eventually help us interpret complex symptoms during and post-infection in humans and have more targeted ways to treat them.