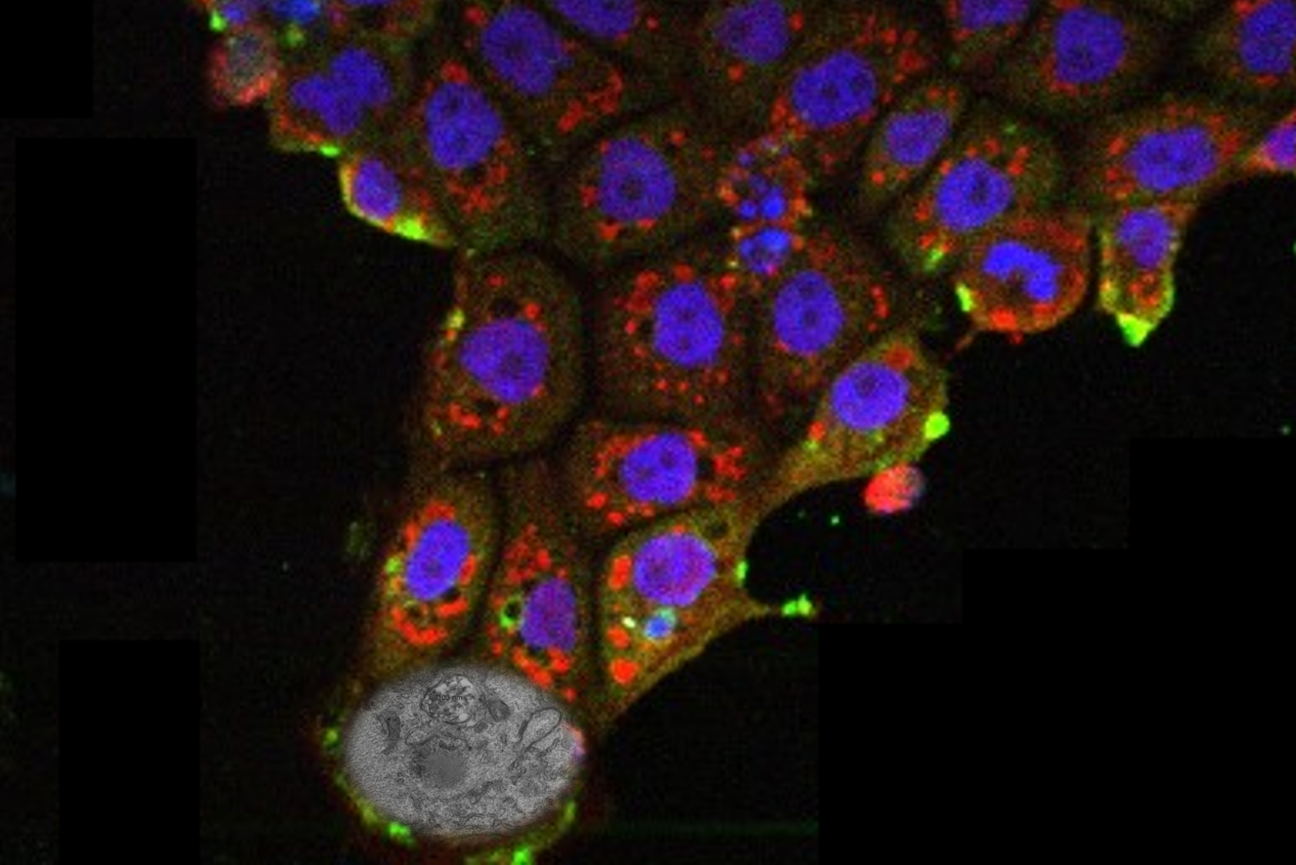
Work led by Talukdar and Page Lab postdoc Lukáš Chmátal shows that there are differences in how healthy male and female heart cells—specifically, cardiomyocytes, the muscle cells responsible for making the heart beat—generate energy.
Greta Friar | Whitehead Institute
February 18, 2025
Heart disease is the number one killer of men and women, but it often presents differently depending on sex. There are sex differences in the incidence, outcomes, and age of onset of different types of heart problems. Some of these differences can be explained by social factors—for example, women experience less-well recognized symptoms when having heart attacks, and so may take longer to be diagnosed and treated—but others are likely influenced by underlying differences in biology. Whitehead Institute Member David Page and colleagues have now identified some of these underlying biological differences in healthy male and female hearts, which may contribute to the observed differences in disease.
“My sense is that clinicians tend to think that sex differences in heart disease are due to differences in behavior,” says Harvard-MIT MD-PhD student Maya Talukdar, a graduate student in Page’s lab. “Behavioral factors do contribute, but even when you control for them, you still see sex differences. This implies that there are more basic physiological differences driving them.”
Page, who is also an HHMI Investigator and a professor of biology at the Massachusetts Institute of Technology, and members of his lab study the underlying biology of sex differences in health and disease, and recently they have turned their attention to the heart. In a paper published on February 17 in the women’s health edition of the journal Circulation, work led by Talukdar and Page lab postdoc Lukáš Chmátal shows that there are differences in how healthy male and female heart cells—specifically, cardiomyocytes, the muscle cells responsible for making the heart beat—generate energy.
“The heart is a hard-working pump, and heart failure often involves an energy crisis in which the heart can’t summon enough energy to pump blood fast enough to meet the body’s needs,” says Page. “What is intriguing about our current findings and their relationship to heart disease is that we’ve discovered sex differences in the generation of energy in cardiomyocytes, and this likely sets up males and females differently for an encounter with heart failure.”
Page and colleagues began their work by looking for sex differences in healthy hearts because they hypothesize that these impact sex differences in heart disease. Differences in baseline biology in the healthy state often affect outcomes when challenged by disease; for example, people with one copy of the sickle cell trait are more resistant to malaria, certain versions of the HLA gene are linked to slower progression of HIV, and variants of certain genes may protect against developing dementia.
Identifying baseline traits in the heart and figuring out how they interact with heart disease could not only reveal more about heart disease, but could also lead to new therapeutic strategies. If one group has a trait that naturally protects them against heart disease, then researchers can potentially develop medical therapies that induce or recreate that protective feature in others. In such a manner, Page and colleagues hope that their work to identify baseline sex differences could ultimately contribute to advances in prevention and treatment of heart disease.
The new work takes the first step by identifying relevant baseline sex differences. The researchers combined their expertise in sex differences with heart expertise provided by co-authors Christine Seidman, a Harvard Medical School professor and director of the Cardiovascular Genetics Center at Brigham and Women’s Hospital; Harvard Medical School Professor Jonathan Seidman; and Zoltan Arany, a professor and director of the Cardiovascular Metabolism Program at the University of Pennsylvania.
Along with providing heart expertise, the Seidmans and Arany provided data collected from healthy hearts. Gaining access to healthy heart tissue is difficult, and so the researchers felt fortunate to be able to perform new analyses on existing datasets that had not previously been looked at in the context of sex differences. The researchers also used data from the publicly available Genotype-Tissue Expression Project. Collectively, the datasets provided information on bulk and single cell gene expression, as well as metabolomics, of heart tissue—and in particular, of cardiomyocytes.
The researchers searched these datasets for differences between male and female hearts, and found evidence that female cardiomyocytes have higher activity of the primary pathway for energy generation than male cardiomyocytes. Fatty acid oxidation (FAO) is the pathway that produces most of the energy that powers the heart, in the form of the energy molecule ATP. The researchers found that many genes involved in FAO have higher expression levels in female cardiomyocytes. Metabolomic data reinforced these findings by showing that female hearts had greater flux of free fatty acids, the molecules used in FAO, and that female hearts used more free fatty acids than did males in the generation of ATP.
Altogether, these findings show that there are fundamental differences in how female and male hearts generate energy to pump blood. Further experiments are needed to explore whether these differences contribute to the sex differences seen in heart disease. The researchers suspect that an association is likely, because energy production is essential to heart function and failure.
In the meantime, Page and his lab members continue to investigate the biology underlying sex differences in tissues and organs throughout the body.
“We have a lot to learn about the molecular origins of sex differences in health and disease,” Chmátal says. “What’s exciting to me is that the knowledge that comes from these basic science discoveries could lead to treatments that benefit men and women, as well as to policy changes that take sex differences into account when determining how doctors are trained and patients are diagnosed and treated.”