The pseudoautosomal region (PAR) is a critical area on the Y chromosome that swaps genetic information with the X chromosome. Recent research from the Page Lab reaffirms the location of PAR and offers a refined understanding of where crossover events occur.
Shafaq Zia | Whitehead Institute
October 14, 2024
At first, the X and the Y sex chromosomes seemed like an unlikely pair. But then, researchers, including Whitehead Institute Member David Page, began finding clues that suggested otherwise: identical DNA sequences on the X and Y chromosomes.
Soon, it became clear that the tips of the X and Y chromosomes join together in a tight embrace, swapping genetic material during the process of sperm production from immature male germ cells. This limited area of genetic exchange between the two sex chromosomes is called the pseudoautosomal region (PAR).
But science is an iterative process—a continuous cycle of questioning, testing, and revising knowledge. Last fall, what had long been considered well established in genetics was called into question when new research suggested that the boundary of the PAR might be half a million base pairs away from the accepted location. Given that a typical human gene is about tens of thousands of base pairs, this length would potentially span multiple genes on the X and Y chromosomes, raising serious concerns about the accuracy and validity of decades of scientific literature.
Fortunately, new work from Page, research scientist Daniel Winston Bellott, and colleagues—published Oct. 14 in the American Journal of Human Genetics—offers clarity. In this study, the group re-examines the size of the PAR using sequencing data presented by outside researchers in their 2023 work, alongside decades of genomic resources, and single-cell sequencing of human sperm. Their findings confirm that the location of the boundary to the PAR, as identified by scientists in 1989, still holds true.
“If one is interested in understanding sex differences in health and disease, the boundary of the pseudoautosomal region is arguably the most fundamental landmark in the genome,” says Page, who is also a professor of biology at the Massachusetts Institute of Technology and an Investigator with Howard Hughes Medical Institute. “Had this boundary been multiple genes off, the field would have been shaken to its foundations.”
Dance of the chromosomes
The X and Y chromosomes evolved from an ancestral pair of chromosomes with identical structures. Over time, the Y chromosome degenerated drastically, losing hundreds of functional genes. Despite their differences, the X and Y chromosomes come together during a special type of cell division called male meiosis, which produces sperm cells.
This process begins with the tips of the sex chromosomes aligning side by side like two strands of rope. As the X and Y chromosomes embrace each other, enzymes create breaks in the DNA. These breaks are repaired using the opposite chromosome as a template, linking the X and Y together. About half of the time, an entire segment of DNA, which often contains multiple genes, will cross over onto the opposite chromosome.
The genetic exchange, called recombination, concludes with the X and Y chromosomes being pulled apart to opposite ends of the dividing cell, ensuring that each chromosome ends up in a different daughter cell. “This intricate dance of the X and Y chromosomes is essential to a sperm getting either an X or a Y—not both, and not neither,” says Page.
This way when the sperm—carrying either an X or a Y—fuses with the egg—carrying an X—during fertilization, the resulting zygote has the right number of chromosomes and a mix of genetic material from both parents.
But that’s not all. The swapping of DNA during recombination also allows for the chromosomes to have the same genes but with slight variations. These unique combinations of genetic material across sex chromosomes are key to genetic diversity within a species, enabling it to survive, adapt, and reproduce successfully.
Beyond the region of recombination, the Y chromosome contains genes that are important for sex determination, for sperm production, and for general cellular functioning. The primary sex-defining gene, SRY, which triggers the development of an embryo into a male, is located only 10,000 bases from the boundary of the PAR.
Advancing together
To determine whether the location of this critical boundary on the human sex chromosomes—where they stop crossing over during meiosis and become X-specific or Y-specific—had been misidentified for over three decades, researchers began by comparing publicly-available DNA sequences from the X and the Y chromosomes of seven primate species: humans, chimpanzees, gorillas, orangutans, siamangs, rhesus macaques, and colobus monkeys.
Based on the patterns of crossover between the X and the Y chromosomes of these species, the researchers constructed an evolutionary tree. Upon analyzing how DNA sequences close to and distant from the PAR boundary group together across species, the researchers found a substitution mutation—where a letter in a long string of letters is swapped for a different one—in the DNA of the human X and Y chromosomes. This change was also present in the chimpanzee Y chromosome, suggesting that the mutation originally occurred in the last common ancestor of humans and chimpanzees and was then transferred to the human X chromosome.
“These alignments between various primates allowed us to observe where the X and the Y chromosomes have preserved identity over millions of years and where they have diverged,” says Bellott. “That [pseudoautosomal] boundary has remained unchanged for 25 million years.”
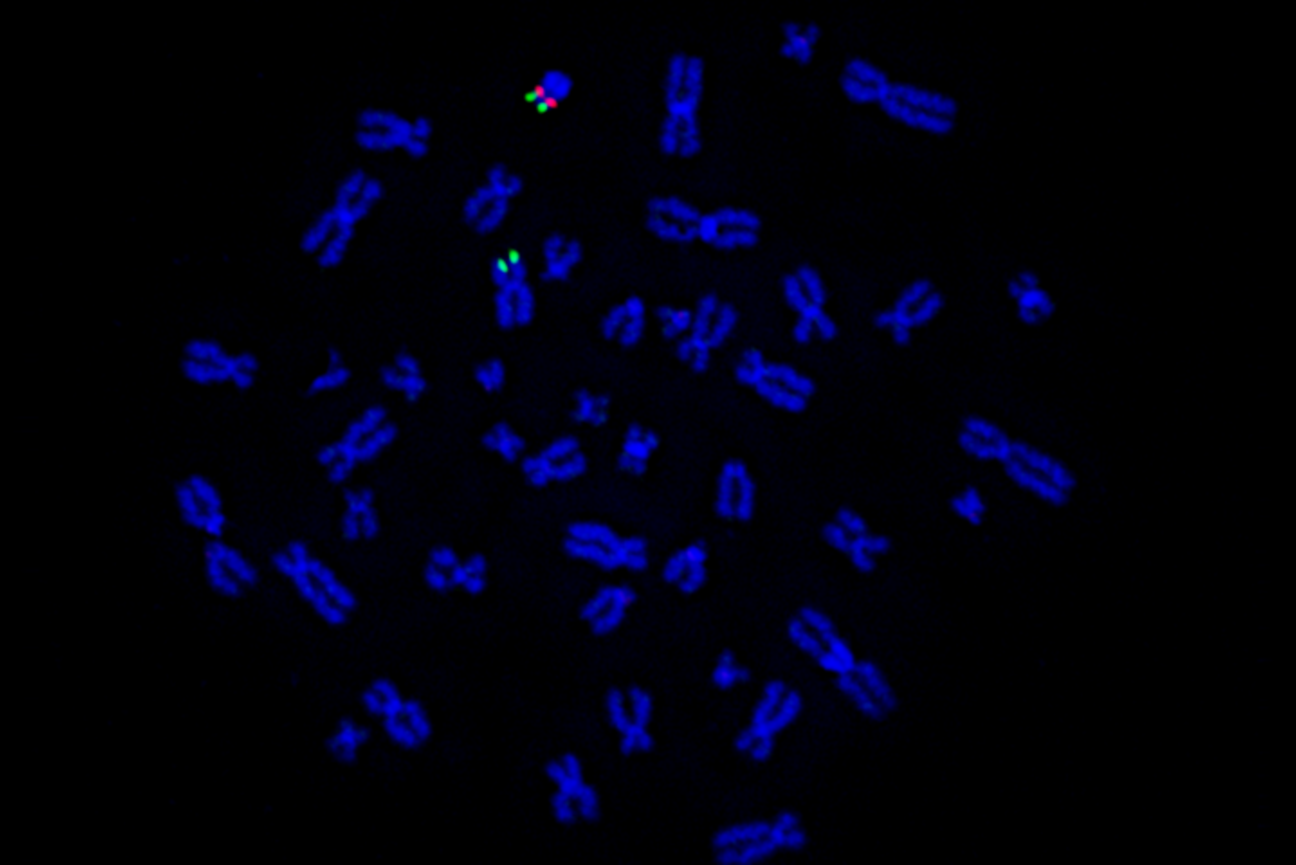
Next, the group studied crossover events in living humans using a vast dataset of single-cell sequencing of sperm samples. They found 795 sperm with clear swapping of genetic material somewhere between the originally proposed boundary of the PAR and the newly-proposed 2023 boundary.
Once these analyses confirmed that the original location of the PAR boundary remains valid, Page and his team turned their attention to data from the 2023 study that contested this 1989 finding. The researchers focused on 10 male genomes assembled by the outside group, which contained contiguous sequences from the PAR.
Since substitutions on the Y chromosome typically occur at a steady rate, but in the PAR, changes on the X chromosome can transfer to the Y through recombination, the researchers compared the DNA sequences from the ten genomes to determine whether they followed the expected steady rate of change or if they varied.
The team found that close to the originally proposed PAR boundary, the DNA sequences changed at a steady rate. But further away from the boundary, the rate of change varied, suggesting that crossover events likely occurred in this region. Furthermore, the group identified several shared genetic differences between the X and the Y chromosomes of these genomes, which demonstrates that recombination has occurred even closer to the PAR boundary than scientists observed in 1989.
“Ironically, instead of contradicting the original boundary, the 2023 work has helped us refine the location of crossover to an even narrower area near the boundary,” says Page.
Thanks to the efforts of Page’s group at Whitehead Institute, our understanding of the PAR is clearer than ever, and business can go on as usual for researchers investigating sex differences in health and disease.