Raleigh McElvery
July 10, 2019
All cells must balance generating new proteins with eliminating excess or damaged ones by way of powerful degradation machines — which, much like wood chippers, chew up proteins and spit them out. But, these proteins are often folded into intricate structures, and must be unfurled before they can be fed into these degradation machines, broken into tiny bits, and ultimately recycled. In bacteria, a molecular motor known as ClpX must grip the end of the ill-fated protein and apply force to straighten it. However, until now, researchers weren’t sure precisely how ClpX gripped its target tightly enough to accomplish this task.
There had been evidence to suggest that some amino acids — the chemical building blocks that comprise proteins — are “slippery” and thus more difficult to grip. In a new study published in eLife, researchers at the MIT Department of Biology examined each amino acid’s individual contribution to grip. By parsing the physical basis for this molecular interaction, they hope to better understand how some proteins evade destruction.
“Previous studies had shown that small amino acids were notoriously hard to grip, but no one really understood why,” says Tristan Bell, graduate student and first author on the paper. “It’s like watching a game of tug-of-war and knowing that a person’s hands are important for pulling on the rope, but having no idea what allows the hands to get a good grip on the rope.”
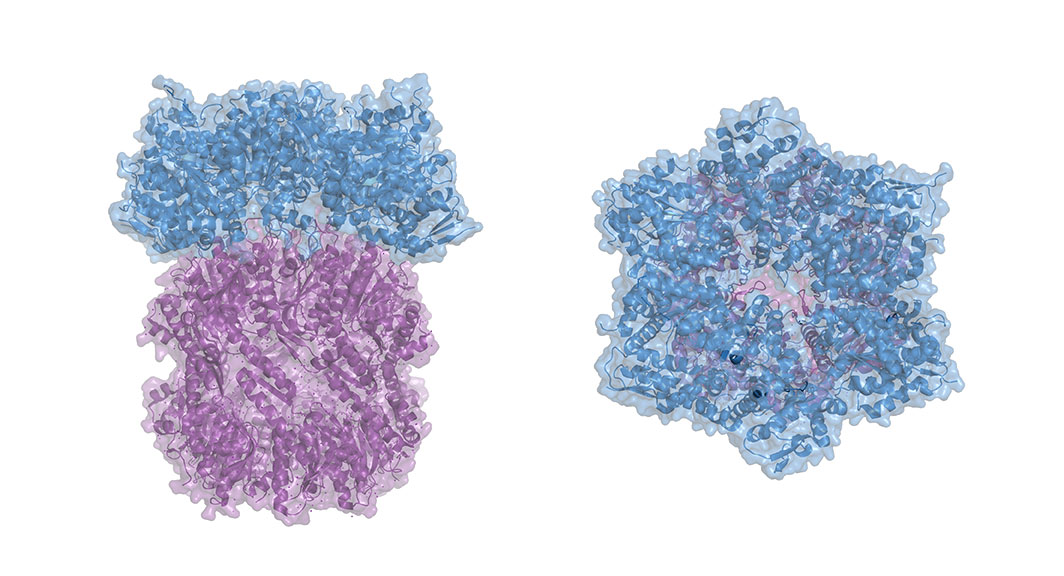
ClpX, he explains, is roughly shaped like donut with loops protruding into the center hole. These loops grip the target protein, jamming it against the surface of ClpX, and unfolding it so it can be threaded through the hole and shredded.
The researchers engineered proteins with tails comprised of various amino acid combinations, and measured how well ClpX could grip them, both in bacteria and in test tubes. They determined that ClpX can only grip between six and eight amino acids at a time, and that only a handful of the 20 possible amino acids could actually be “well-gripped.” When ClpX was able to grasp multiple amino acids simultaneously, its grip strength increased.
“We think that somehow the charge is preventing ClpX from making strong contacts with the target protein, preventing it from achieving a stable grip state,” Bell says.Just like in previous experiments, large amino acids appeared easier to grip than small ones, “similar to the way a knotted rope is easier to grasp than a smooth, slippery one,” Bell says. But, regardless of size, amino acids that carried electric charge seemed to be more slippery.
The team thinks that proteins with slippery tails might have an evolutionary advantage, because they are harder to grip and therefore less likely to be degraded.
Invaders like viruses have been known to insert a slippery sequence into certain proteins to prevent the host cell from destroying them and thus promoting replication. Even healthy cells produce proteins with strategically placed slippery sequences, which allow a portion of the protein to break away from the degradation machinery unscathed. In the bacteria Caulobacter crescentus, this planned breakage actually produces a version of one protein that’s needed for DNA replication.
“Next,” Bell says, “we’re hoping to look across entire proteomes in different organisms to find more proteins that escape destruction.”
“Tristan’s experiments and results reveal some of the molecular determinants of grip in the bacterial degradation machines we study,” says Bob Sauer, the Salvador E. Luria Professor of Biology and senior author on the study. “Many of the rules he discovered apply to related machines that function in all biological organisms, including humans, emphasizing the common evolution of these machines.”
Citation:
“Interactions between a subset of substrate side chains and AAA+ motor pore loops determine grip during protein unfolding”
eLife, online June 28, 2019, DOI: 10.7554/eLife.46808
Tristan A. Bell, Tania A. Baker, and Robert T. Sauer.