MIT senior Daniel Zhang aims to provide hope for young patients and support to young students.
Celina Zhao | Department of Biology
February 24, 2022
During the virtual spring 2020 semester, Daniel Zhang, a senior majoring in biology, put his time at home to good use. In the garage of his home in San Diego, California, Zhang helped his 13-year-old brother build a lab to study dry eye disease.
This combination of mentorship and medicine feels like second nature to Zhang. When his parents opened a family-run optometry clinic, Zhang was their first patient and then their receptionist. And after a close family member passed away from leukemia, he remembers thinking, “Humans are susceptible to so many diseases — why don’t we have better cures?”
That question propelled him to spend his high school summers studying biomarkers for the early detection of leukemia at the University of California at San Diego. He was invited to present his research at the London International Youth Science Forum, where he spoke to scientists from almost 70 countries. Afterward, he was hooked on the idea of scientific research as a career.
“Research is like standing on the shoulders of giants,” he says. “My experience at the forum was when I knew I loved science and wanted to continue using it to find common ground with others from completely different cultures and backgrounds.”
Exploring the forefront of cancer research
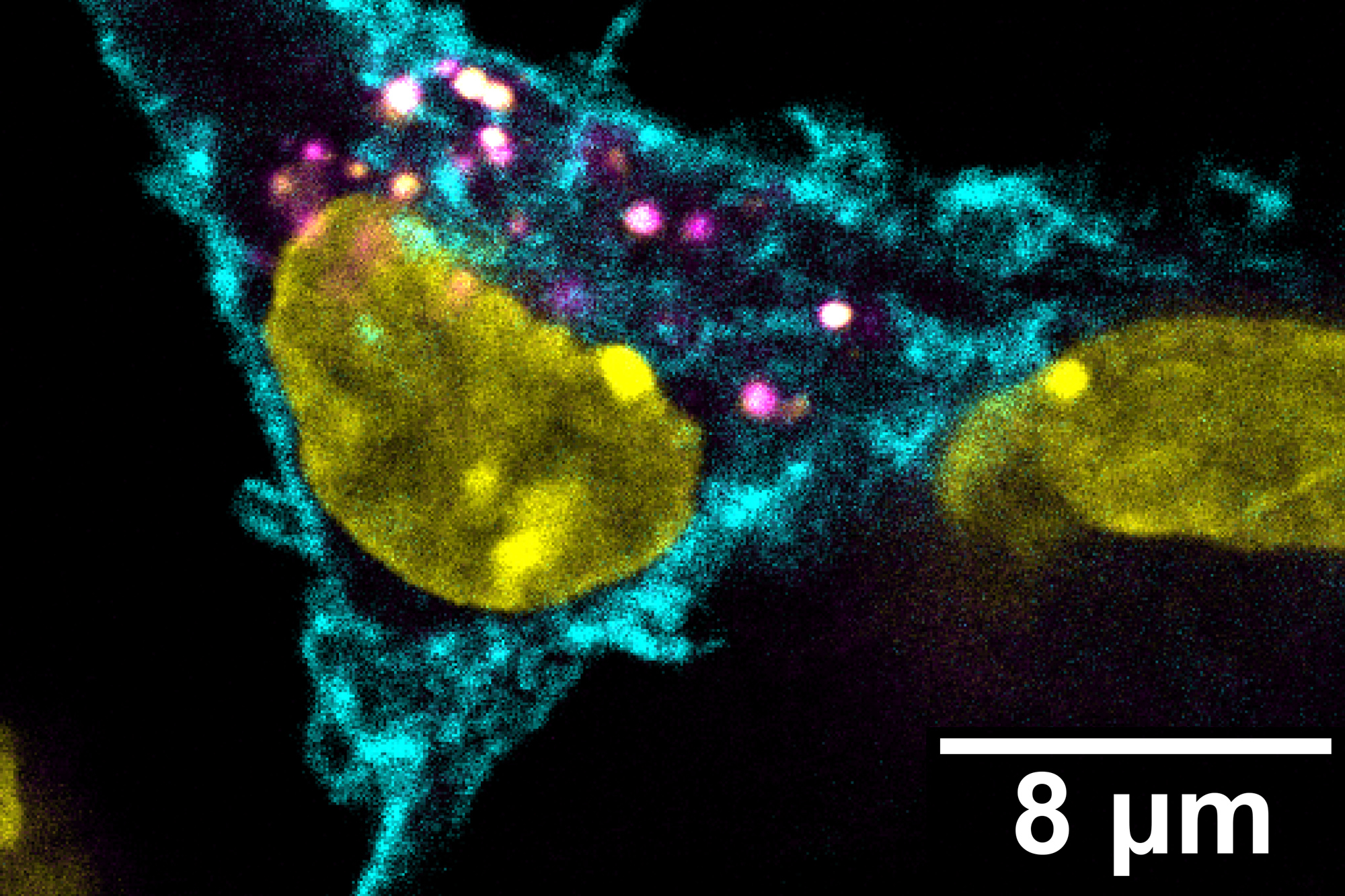
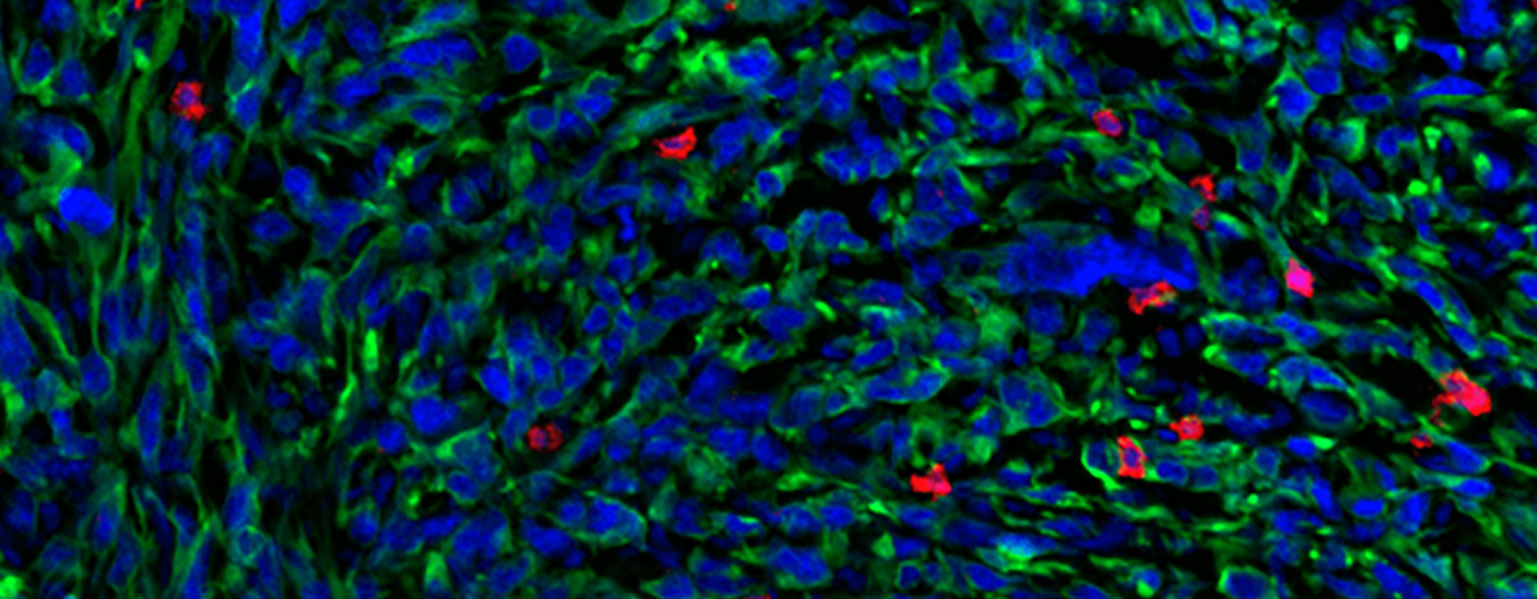
As soon as he arrived at MIT as a first-year undergraduate, Zhang began working under the guidance of postdoc Peter Westcott in professor Tyler Jacks’ lab. The lab focuses on developing better mouse and organoid models to study cancer progression — in Zhang’s case, metastatic colorectal cancer.
One of the ways to model colorectal cancer is by injecting an engineered virus directly into the colons of mice. The viruses, called lentiviral agents, “knock out” tumor suppressor genes and activate the so-called oncogenes that drive cancer forward. However, the imprecise nature of this injection also unintentionally transforms many “off-target” cells into cancer cells, producing a cancer that’s far too widespread and aggressive. Additionally, rare tumors called sarcomas are often initiated rather than adenocarcinomas, the type of tumor found in 95 percent of human cases. As a result, these mouse models are limited in their ability to accurately model colorectal cancer.
To address this problem, Zhang and Westcott designed a method using CRISPR/Cas9 to target a special stem cell called LGR5+, which researchers believe are the types of cells that, when mutated, grow into colorectal cancer. His technique modifies only the LGR5+ cells, which would allow researchers to control the rate at which adenocarcinomas grow. Therefore, it generates a model that is not only much more similar to human colorectal cancer than other models, but also allows researchers to quickly test for other potential cancer driver genes with CRISPR/Cas9. Designing an accurate model is crucial for developing and testing effective new therapies for patients, Zhang says.
During MIT’s virtual spring and fall semesters of 2020, Zhang shifted his focus from benchwork in the lab to computational biology. Using patient data from the Cancer Genome Atlas, Zhang analyzed mutation rates and discovered three genes potentially involved in colorectal cancer tumor suppression. He plans to test their function in his new mouse model to further validate how the dysfunction of these genes drives colorectal cancer progression.
For his work on organoid modeling of colorectal cancer, a third project he’s worked on during his time at the Jacks lab, he also won recognition from the American Association for Cancer Research (AACR). As one of 10 winners of the Undergraduate Scholar Award, he had the opportunity to present his research at the virtual AACR conference in 2021 and again at the next AACR Conference in New Orleans in April 2022.
He credits MIT’s “mens et manus” philosophy, encouraging the hands-on application of knowledge, as a large part of his early success in research.
“I’ve found that, at MIT, a lot of people are pursuing projects and asking questions that have never been thought of before,” Zhang says. “No one has ever been able to develop a late-stage model for colorectal cancer that’s amenable to gene editing. As far as I know, other than us, no one in the world is even working on this.”
Inspiring future generations to pursue STEM
Outside of the lab, Zhang devotes a substantial amount of time to sharing the science he’s so passionate about. Not only has he been awarded the Gene Brown Prize for undergraduate teaching for his time as a teaching assistant for the lab class 7.002 (Fundamentals of Experimental Molecular Biology), but he’s also taken on leadership roles in science outreach activities.
During the 2020-21 academic year, he served as co-director of DynaMIT, an outreach program that organizes a two-week STEM program over the summer for underserved sixth to ninth graders in the greater Boston area. Although the program is traditionally held in-person, in summer 2021 it was held virtually. But Zhang and the rest of the board didn’t let the virtual format deter them from maximizing the fun and interactive nature of the program. They packed and shipped nearly 120 science kits focused on five major topics — astronomy, biology, chemistry, mechanical engineering, and math — allowing the students to explore everything from paper rockets to catapults and trebuchets to homemade ice cream.
“At first, we were worried that most of the students wouldn’t turn on their cameras, since we saw that trend all over MIT classes during the semester,” Zhang says. “But almost everyone had their cameras on the entire time. It was really gratifying to see students come in on Monday really shy, but by Friday be actively participating, making jokes with the mentors, and being really excited about STEM.”
To investigate the long-term impacts of the program, he also helped kick-start a project that followed up with DynaMIT alumni, some of whom have already graduated from college. Zhang says: “We were happy to see that 80-90 percent of DynaMIT alumni enjoyed the program, rating it four or five out of five, and close to 70 percent of them said that DynaMIT had a really positive impact on their trajectory toward a career in STEM.”
Zhang has also served as president of the MIT Pre-medical Society, with the goals of fostering an encouraging environment for premed undergraduates, and providing guidance and resources to first- and second-year students still undecided about the premed path. To achieve these objectives, he pioneered an MIT-hosted mixer with the premedical societies of other Boston colleges, including Wellesley College, Boston University, Tufts University, and Harvard University. At the mixer, students were able to network with each other and listen to guest speakers from the different universities talk about their experiences in medicine. He also started a “big/little” initiative that paired third- and fourth-year mentors with first- and second-year students.
Providing new opportunity and hope
The wealth of activities Zhang has participated in at MIT has inspired his choices for the future. After graduation, he plans to take a gap year and work as a research technician in pediatric oncology before applying to MD/PhD programs.
On the mentorship side, he’s currently working to establish a nonprofit organization called Future African Scientist with his former Ugandan roommate, Martin Lubowa, whom he met at a study abroad program during MIT’s Independent Activities Period in 2020. The organization will teach high schoolers in Africa professional skills and expose them to different STEM topics — a project Zhang plans to work on post-MIT and into the long term.
Ultimately, he hopes to lead his own lab at the intersection of CRISPR-Cas9 technology and cancer biology, and to serve as a mentor to future generations of researchers and physicians.
As he puts it: “All of the experiences I’ve had so far have solidified my goal of conducting research that impacts patients, especially young ones. Being able to provide new opportunity and hope to patients suffering from late-stage metastatic diseases with no current cures is what inspires me every day.”