Greta Friar | Whitehead Institute
January 4, 2023
Many cancer cells never leave their original tumors. Some cancer cells evolve the ability to migrate to other tissues, but once there cannot manage to form new tumors, and so remain dormant. The deadliest cancer cells are those that can not only migrate to, but also thrive and multiply in distant tissues. These metastatic cancer cells are responsible for most of the deaths associated with cancer. Understanding what enables some cancer cells to metastasize—to spread and form new tumors—is an important goal for researchers, as it will help them develop therapies to prevent or reverse those deadly occurrences.
Past research from Whitehead Institute Member Robert Weinberg and others suggests that cancer cells are best able to form metastatic tumors when the cells are in a particular state called the quasi-mesenchymal (qM) state. New research from Weinberg and Arthur Lambert, once a postdoc in Weinberg’s lab and now an associate director of translational medicine at AstraZeneca, has identified two gene-regulating molecules as important for keeping cancer cells in the qM state. The research, published in the journal Developmental Cell on December 19, shows that these molecules, ΔNp63 and p73, enable breast cancer cells to form new tumors in mice, and illuminates important aspects of how they do so.
Most potent in the middle
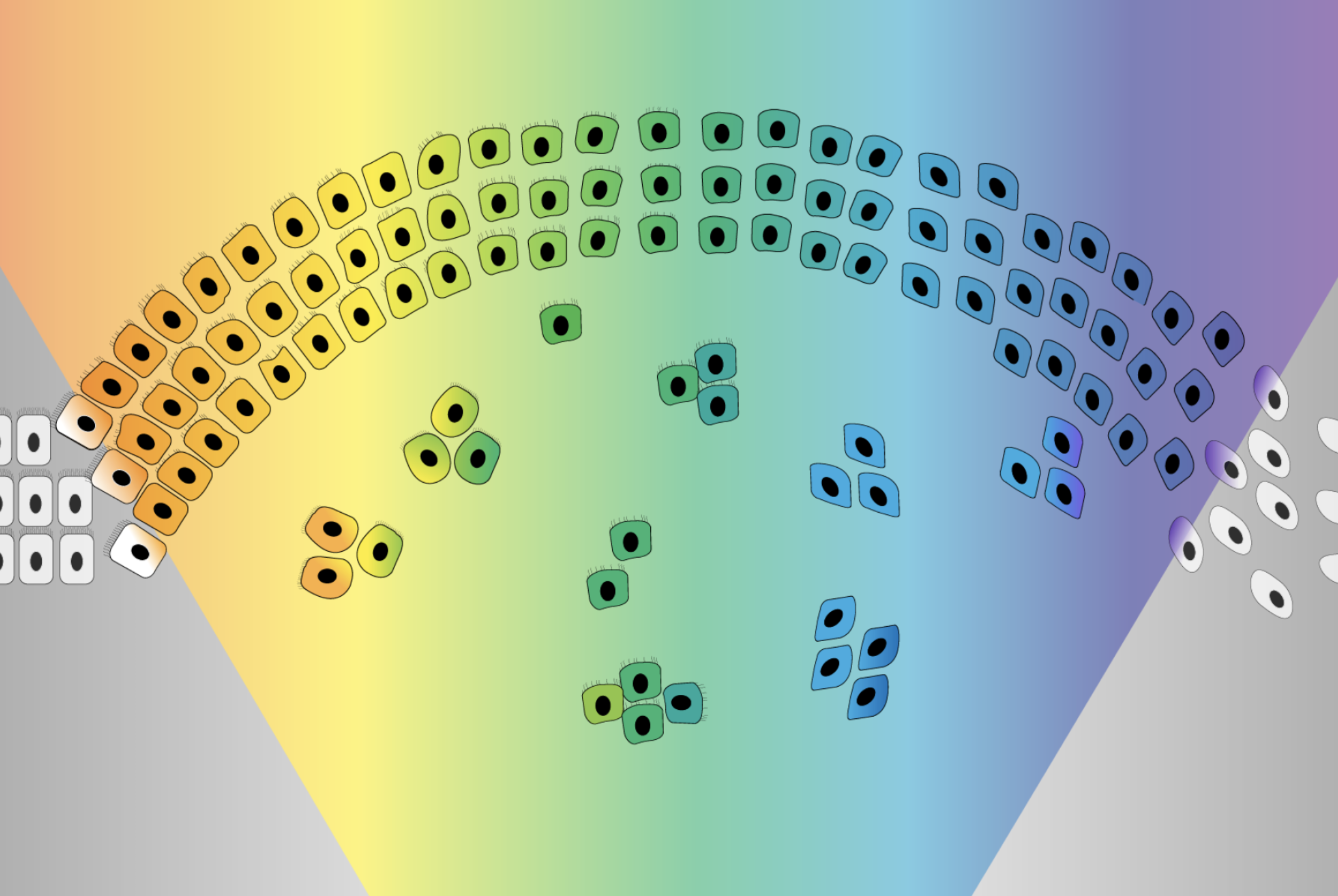
Cells enter the qM state by undergoing the epithelial-mesenchymal transition (EMT), a developmental process that can be co-opted by cancer cells. In the EMT, cells transition from an epithelial state through a spectrum of more mesenchymal states, which allows them to become more mobile and aggressive. Cells in the qM state have only transitioned partway through the EMT, becoming more, but not fully, mesenchymal. This middle ground is perfect for metastasis, whereas cells on either end of the spectrum—cells that are excessively epithelial or excessively mesenchymal—lose their metastatic abilities.
Lambert and colleagues wanted to understand more about how cancer stem cells, which can seed metastases and recurrent tumors, remain in a metastasis-prone qM state. They analyzed how gene activity was regulated in those cells and identified two transcription factors—molecules that influence the activity of target genes—as important. One of the transcription factors, ΔNp63, appeared to most directly control cancer stem cells’ ability to maintain a qM state. The other molecule, p73, seemed to have a similar role because it can activate ΔNp63. When either transcription factor was inactivated, the cancer stem cells transitioned to the far end of the EMT spectrum and so were unable to metastasize.
Next, the researchers looked at what genes ΔNp63 regulates in cancer stem cells. They expected to find a pattern of gene regulation resembling what they would see in healthy breast stem cells. Instead they found a pattern closely resembling what one would see in cells involved in wound healing and regeneration. Notably, ΔNp63 stimulates EGFR signaling, which is used in wound healing to promote rapid multiplication of cells.
“Although this is not what we expected to see, it makes a lot of sense because the process of metastasis requires active proliferation,” Lambert says. “Metastatic cancer cells need both the properties of stem cells—such as the ability to self-renew and differentiate into different cell types—and the ability to multiply their numbers to grow new tumors.”
This finding may help to explain why qM cells are so uniquely good at metastasizing. Only in the qM state can the cells strongly stimulate EGFR signaling and so promote their own proliferation.
“This work gives us some mechanistic understanding of what it is about the quasi-mesenchymal state that drives metastatic tumor growth,” says Weinberg, who is also the Daniel K. Ludwig Professor for Cancer Research at the Massachusetts Institute of Technology.
The researchers hope that these insights could eventually contribute to therapies that prevent metastasis. They also hope to pursue further research into the role of ΔNp63. For example, this work illuminated a possible connection between ΔNp63 and the activation of dormant cancer cells, the cells that travel to new tissues but then cannot proliferate after they arrive there. Such dormant cells are viewed as ticking time bombs, as at any point they may reawaken. Lambert hopes that further research may reveal new insights into what causes dormant cancer cells to eventually gain the ability to grow tumors, adding to our understanding of the mechanisms of metastatic cancer.
Notes
Arthur W. Lambert, Christopher Fiore, Yogesh Chutake, Elisha R. Verhaar, Patrick C. Strasser, Mei Wei Chen, Daneyal Farouq, Sunny Das, Xin Li, Elinor Ng Eaton, Yun Zhang, Joana Liu Donaher, Ian Engstrom, Ferenc Reinhardt, Bingbing Yuan, Sumeet Gupta, Bruce Wollison, Matthew Eaton, Brian Bierie, John Carulli, Eric R. Olson, Matthew G. Guenther, Robert A. Weinberg. “ΔNp63/p73 drive metastatic colonization by controlling a regenerative epithelial stem cell program in quasi-mesenchymal cancer stem cells.” Developmental Cell, Volume 57, Issue 24,
2022, 2714-2730.e8, https://doi.org/10.1016/j.devcel.2022.11.015.