Greta Friar | Whitehead Institute
April 5, 2019
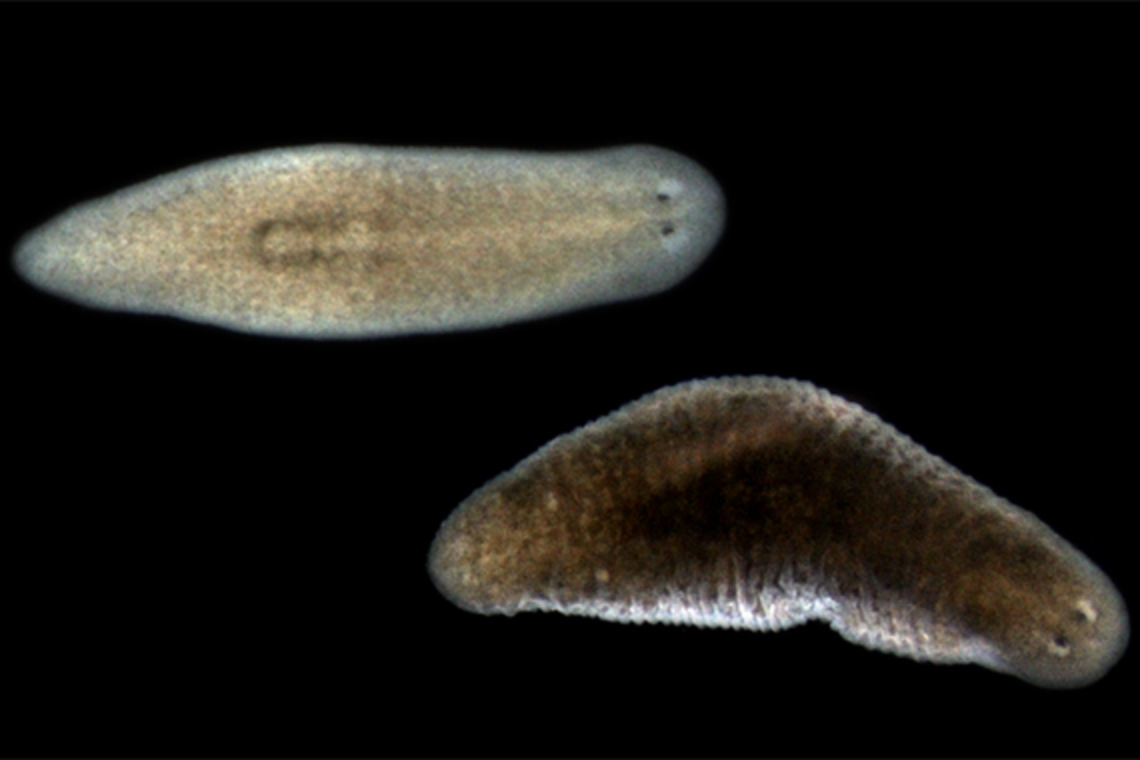
CAMBRIDGE, MA — Planarians are flatworms best known for their incredible ability to regenerate all their body parts: chop a planarian in two and soon you will have two perfectly formed planarians. As Whitehead Institute Member Peter Reddien, also a professor of biology at Massachusetts Institute of Technology and an investigator with the Howard Hughes Medical Institute, has investigated planarians over the years, he has become increasingly fascinated with the functions of their muscle. Not only do planarians use muscle to move, but Reddien’s research group previously discovered they rely on muscle tissue to provide a full body map with instructions that helps guide stem cells to the right locations during both regeneration and normal turnover of cells. Muscle tissue does this by secreting positional signals that help cells identify where they are — and where they should be.
New research from Reddien and graduate student Lauren Cote shows that muscle serves yet another crucial function in planarians. In a paper published in Nature Communications on April 8, they show that muscle operates as the planarian’s connective tissue, providing basic architectural support for the body. Connective tissue functions in large part by secreting molecules that make up the extracellular matrix (ECM), a network of molecules outside of the body’s cells that provides tissues with, among other things, scaffolding, protection, separation of tissues, and a means of inter-tissue connection and communication. In vertebrates, including humans, connective tissue is a distinct tissue type containing dedicated cells such as fibroblasts that secrete most of the animal’s ECM proteins. Reddien and Cote found no such fibroblast-like cell type in planarians; instead, multipurpose muscle does it all.
The researchers began to suspect that planarian muscle might function as connective tissue when they discovered that the gene encoding a major type of ECM molecule, fibrous protein collagen, was expressed only in muscle. The researchers then catalogued the total collection of proteins found in the planarian’s ECM, called the matrisome, and tracked where the genes that code for those proteins were expressed. They identified nineteen collagen genes, and all nineteen were highly specific to muscle. The vast majority of other ECM genes followed suit.
To further test muscle’s role as connective tissue, the researchers silenced the gene hemicentin-1, which produces another ECM molecule expressed specifically in muscle. They found that when the gene was not expressed, the planarian’s inner tissues did not remain properly separated from its outer skin. In other words, a muscle-specific gene is necessary in the planarians they studied for the core connective tissue task of keeping tissues discrete.
Although it might seem unusual that planarians would use muscle tissue for both ECM secretion and body pattern maintenance, Reddien and Cote say the combination makes a certain sense.
“To establish a map of the body, muscle secretes positional signals, and in its role as connective tissue it is simultaneously creating the extracellular environment the signals travel through,” Reddien says.
Cote agrees: “Producing the body’s physical architectural support and its biochemical architectural blueprint seem to go hand in hand.”
One possibility raised by this synchronicity is that a link between connective tissue and harboring positional information exists broadly across animal species. Studies elsewhere have found some positional role or positional memory in connective tissues in several species, including axolotls, vertebrates capable of limb regeneration. Based on these observations, Reddien says, it would be interesting to consider the positional role that connective tissue cells, like fibroblasts, might play in humans and might have in instructing regeneration broadly.
Written by Greta Friar
***
Peter Reddien’s primary affiliation is with Whitehead Institute for Biomedical Research, where his laboratory is located and all his research is conducted. He is also a Howard Hughes Medical Institute Investigator and a professor of biology at Massachusetts Institute of Technology. The authors also acknowledge the Eleanor Schwartz Charitable Foundation for support.
***
Full citation:
“Muscle functions as a connective tissue and source of extracellular matrix in planarians”
Nature Communications, online April 8, 2019. DOI: 10.1038/s41467-019-09539-6
Lauren E. Cote, Eric Simental, and Peter W. Reddien.