
Two treatment methods enhance chemotherapy by the same means: cellular senescence
Raleigh McElvery
In 2019, cancer researchers from MIT found a drug that targeted an elusive molecular interaction previously considered “undruggable.” In so doing, they opened up a new realm of chemotherapy. This drug, called JH-RE-06, sensitized tumors to treatment, but the scientists didn’t fully understand how it exerted its effects. Now, in a pair of studies published in PNAS, the same team is closer to determining which cellular processes this drug alters to enhance cancer therapy.
Many widely-used chemotherapies, like cisplatin, kill tumors by damaging their DNA and inducing programmed cell death. But cells are resilient, and many can continue to function with the help of repair enzymes that simply bypass the damage and continue to replicate the DNA. As a result, some tumors not only defy death, but gain mutations that render them more resistant to treatment or spur new malignancies elsewhere.
“If you’re not making a patient better, it’s very likely you’re making them worse,” says Michael Hemann, associate professor of biology and co-author on both studies. Rather than fully replacing conventional DNA-damaging treatments — which could take decades — Hemann suggests an effective “medium-term” solution: augmenting low doses of cisplatin with safer agents that strengthen the chemotherapy’s tumor-killing capacity.
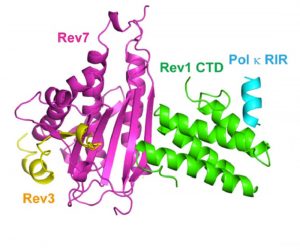
The team had tested this approach back in 2019, when they first identified JH-RE-06 and saw it enhanced chemotherapy treatment. These experiments revealed that JH-RE-06 bound to an especially shallow (and infamously undruggable) pocket of one DNA repair enzyme called REV1. This barred REV1 from interacting with another key enzyme, and prevented the cancer cells from recovering after cisplatin treatment. But what happened next to cause the tumors to shrink was unclear.
As they began their next round of experiments, the researchers expected to find that the drug would simply enhance the way cisplatin kills tumors via programmed cell death.
“We thought that if we blocked the DNA repair process with the JH compound, we’d see more programmed cell death,” says co-author Graham Walker, American Cancer Society Professor and Howard Hughes Medical Institute Professor. “As it turns out, we did see more cell death — just not the kind we were expecting.”
Nimrat Chatterjee, Walker’s former postdoc and lead author of the first study, treated mice and individual cells with a combination of cisplatin and JH-RE-06. She expected to see signs of programmed cell death, but for months, she saw no such markers.
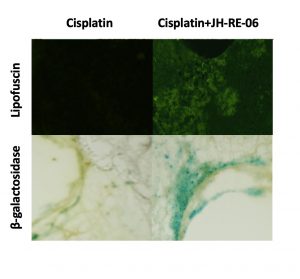
One evening, just as she was about to head home for the day, she peeked through her microscope at the cancer cells treated with JH-RE-06 and cisplatin. She noticed they were fluorescing a strange green color.
“At first, I didn’t know what I was seeing,” she recalls. But after some follow-up, it became clear that the mysterious green color was coming from lipid-containing residues that usually appear as cells age and stop dividing. The cells appeared to be in a permanently dormant state known as senescence — not yet dead but unable to proliferate. JH-RE-06 was altering cisplatin function by triggering a second molecular pathway independent of programmed cell death.
“That was one of the best ‘aha’ moments of my scientific career so far,” Chatterjee says. “REV1, the DNA repair enzyme that JH-RE-06 binds, may have other novel biological functions and a larger role in cancer cell etiology than we originally thought. We’re now grappling with more questions about REV1 than ever before.”

Around the same time, Faye-Marie Vassel PhD ’20, Walker and Hemann’s former joint graduate student and lead author of the second study, witnessed a similar phenomenon in her own experiments. She was investigating a different way of inhibiting the two key DNA repair enzymes that enable cancer cells to survive chemotherapy. Instead of probing JH-RE-06, which latches onto REV1, she tried deleting REV1’s binding partner, called REV7. This protein is particularly influential because it serves an important role in fixing double-stranded breaks in addition to interacting with REV1.
When Vassel deleted REV7 from mice with non-small cell lung cancer, the tumors became more sensitive to cisplatin, as expected. But, like Chatterjee, she saw signs of senescence rather than programmed cell death. The two studies had converged on a common biology: adding JH-RE-06 or deleting REV7 strengthened the effects of cisplatin by inducing this dormant state.
Cancer detection and treatment methods have improved dramatically in the last two decades, but drug-resistant cancers like non-small cell lung cancer remain difficult to combat, Vassel says. “Our experiments are the first to show that senescence induction is likely a consequence of REV7 inhibition,” she adds. “Inhibiting REV7 in tandem with cisplatin therapy may prove to be an effective strategy for enhancing a chemotherapeutic response.”
Chemotherapies that trigger programmed cell death have been the mainstay of cancer treatment for decades. But studies like these show that triggering senescence may be a promising complementary strategy. Most senescent cells are eventually cleared by the immune system, and the researchers suspect this is how cancer cells treated with JH-RE-06 or REV7 inhibitors would be eliminated from the body.
Walker and Hemann agree that, at the moment, their sister studies raise more questions than answers. As Walker explained, “We’ve pried open a new discovery, and hopefully set the stage for many exciting experiments to come.”
Citations:
“REV1 inhibitor JH-RE-06 enhances tumor cell response to chemotherapy by triggering senescence hallmarks”
Proceedings of the National Academy of Sciences, online November 9, 2020, DOI: 10.1073/pnas.2016064117
Nimrat Chatterjee , Matthew A Whitman, A Harris , Sophia M Min , Oliver Jonas , Evan C Lien , Alba Luengo , Matthew G Vander Heiden , Jiyong Hong , Pei Zhou , Michael T Hemann , and Graham C Walker
“Rev7 loss alters cisplatin response and increases drug efficacy in chemotherapy-resistant lung cancer”
Proceedings of the National Academy of Sciences, online November 3, 2020, DOI: 10.1073/pnas.2016067117
Faye-Marie Vassel, Ke Bian, Graham C. Walker, and Michael T. Hemann