Despite being 2,000 miles apart, two researchers are devising deep learning algorithms to predict embryonic tissue folding.
Raleigh McElvery
May 18, 2020
Since March when MIT’s new COVID-19 policies took effect, the research labs on campus have been vacant, save a skeleton crew of essential workers. Despite being separated from their benches, microscopes, and pipets, biologists have devised creative solutions to continue working remotely. In one lab, a postdoc and an undergraduate are using their time at home to develop a deep learning algorithm to spot hidden clues about embryonic development.
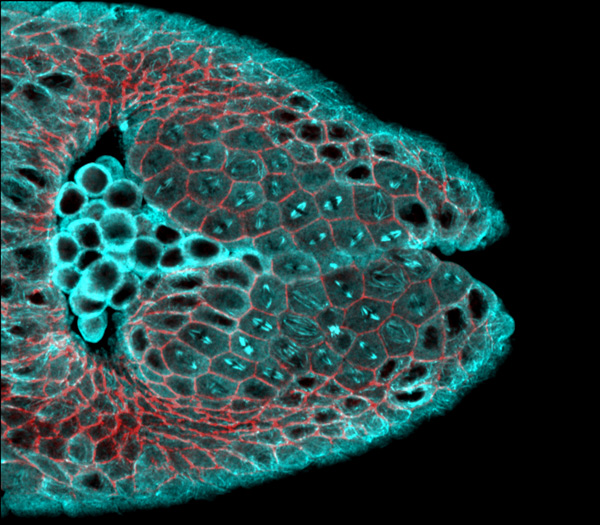
Professor Adam Martin’s lab studies the fruit fly embryo, which consists of a single layer of cells encircling a yolk core about three hours after fertilization. Within the next few minutes, a band of cells on the surface furrows inward, forming a critical fold that helps determine where the cells will go and what roles they will eventually play.
Postdoc Hannah Yevick has spent most of her time in the Martin lab focusing on the protein myosin, which forms a network of connections that links cells together and helps generate the force needed to fold the embryo. With her eye to the microscope, she’s been investigating how this ball of cells compensates for damage and continues to fold correctly despite occasional disruptions to the myosin network. But it remains unclear how cells coordinate to overcome such impediments, and what factors besides myosin aid the process. Yevick began to wonder if there was a way to extract hidden clues from her microscope pictures that would predict which embryos would develop properly and which would not.
Deep learning, a type of machine learning, has become a popular tool to detect and classify visual data. Just like the brain, deep learning algorithms run on sets of interconnecting nodes that can be trained to distinguish features and predict outcomes. (For example, differentiating a cat from a dog, or recognizing a friend in a Facebook picture.) Before an algorithm can complete these tasks on its own, however, researchers must train it using a set of practice images. Some scientists are training algorithms intended for use in clinical settings, from AI-based chatbots to diagnostic assistance that helps predict whether a patient has cancer.

“Deep learning shows great promise in clinical settings,” Yevick says, “and that got me thinking about ways to bring it back into the lab, and dig deeper into fundamental questions about development.”
Although she conducts computational analyses to decipher her microscopy images of fly embryos, Yevick hadn’t considered leveraging deep learning algorithms to predict developmental outcomes until a few months ago. In fact, she’d never tried any machine learning techniques at all. Sitting at home sans microscope during a pandemic seemed like the perfect time to start.
Right before the Martin lab dispersed per MIT’s COVID-19 policies, Yevick gained a collaborator: undergraduate researcher Prateek Kalakuntla, a third-year Course 20 (Biological Engineering) major with a minor in Course 6 (Electrical Engineering and Computer Science). He returned to his home in Dallas, Texas while Yevick remained in Cambridge.
“I was looking for a new project, and this seemed like the perfect one to start from home,” Kalakuntla says. “Our experience of practical machine learning is limited, so we assign ourselves research to do individually, and then check in with each other regularly.”
Despite nearly 2,000 miles separating them, the duo meets via Zoom once or twice a week to discuss their progress. They have been taking online tutorials in deep learning, provided by MIT OpenCourseWare, and gleaning information from scientific papers and colleagues.
“When you’re learning new things, it’s fun to have someone else to bounce ideas off,” Yevick says. “We’re exploring machine learning and gaining basic skills that will help us shape and address important questions moving forward.”

At the moment, they’re practicing by constructing codes pulled from online exercises. Eventually, they aim to create and train their own algorithm and feed it images of embryos, taken just a few minutes into the stage of development where the layer of cells begins to furrow inward. The algorithm will then predict whether or not the embryo will develop correctly over the course of the 15-minute folding process.
Yevick and Kalakuntla intend to collect images from the entire lab, gathering as much data as possible to teach the algorithm to discern successful folds from failed ones. But they hope the algorithm will eventually teach them a thing or two as well — namely, where and when critical proteins are working to influence development.
“We’re feeding the algorithm entire images, but it’s pulling out what it deems to be the most interesting parts,” Kalakuntla says. “These could be specific regions of tissue or time periods that provide hints about the necessary proteins and cell shapes, which we can then analyze further.”
Although they’ll train their algorithm on images of fruit fly embryos, Kalakuntla hopes their model could eventually be applied to other organisms like mice or frogs — and even predict outcomes for data sets lacking images of later developmental stages.
“Machine learning can give us a birds-eye view of how cells coordinate collective movements, and show us ‘signatures’ that we might not have otherwise considered,” Yevick says. “Working remotely is certainly not ideal, but it’s given us the chance to gain new skills like this.”