Raleigh McElvery
November 4, 2019
A new study from the MIT Department of Biology suggests we may need to re-think how certain RNAs operate to impact development and disease.
According to the “central dogma” of biology, DNA is converted into messenger RNA (mRNA) before being expressed as a protein. However, not all RNAs are destined to become proteins. MicroRNAs (miRNAs) are small, non-coding RNAs, which regulate a variety of cellular processes by binding to mRNAs and destabilizing them to reduce their expression.
A single miRNA can target hundreds of different mRNAs. And yet, on its own, an individual miRNA only represses the expression of each mRNA target by about 10-20%. Given that the effects of a single miRNA are so mild, researchers couldn’t understand how they could exert such powerful control over so many processes. One theory is that, rather than acting alone, perhaps multiple miRNAs bind to the same target mRNA in concert to exert enhanced repression. However, few studies have explored this idea in-depth, or identified examples of such co-regulation.
In a new study published in Genome Research on October 24, MIT biologists were able to pinpoint specific miRNAs that collaborate with one another to repress mRNA expression in the brain — adding credence to the notion that miRNAs often collaborate with one another.
“The idea that miRNAs may work by co-targeting sets of transcripts together has been around for a while,” says Jennifer Cherone, the study’s lead author. “But it’s only recently that certain key advances — like better annotations of where transcripts end and more accurate predictions of miRNA target sites — have allowed us to uncover these relationships and rigorously test them in the lab.”
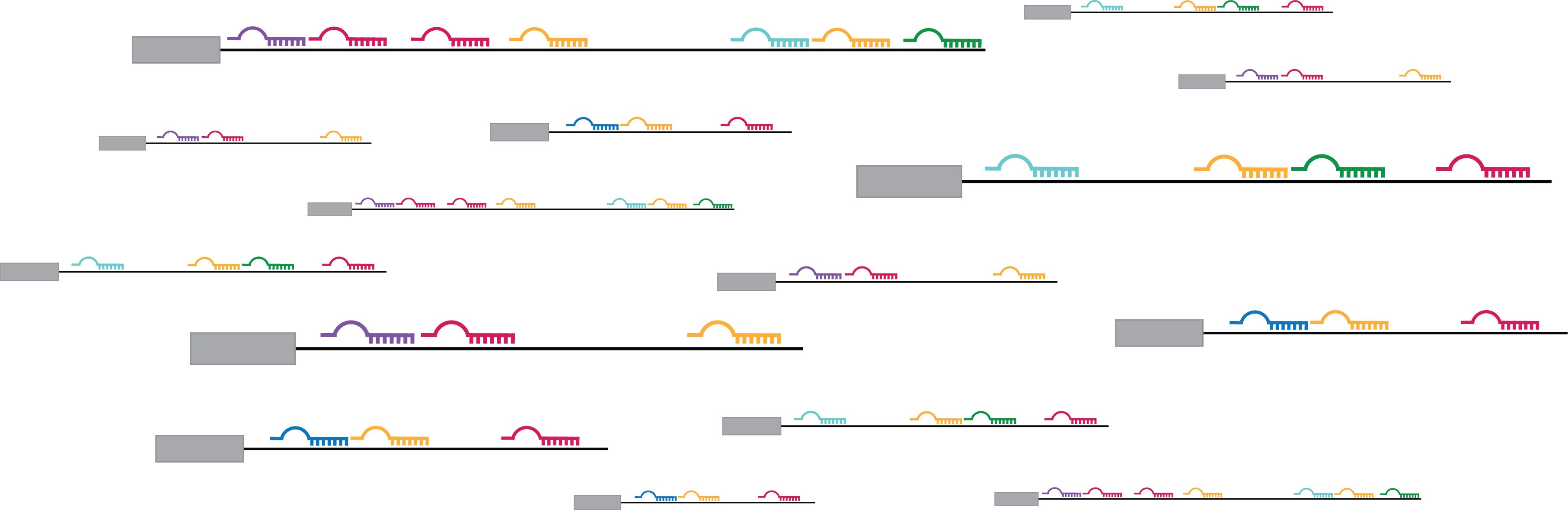
Using powerful computational analyses to compare target sets of different miRNAs, Cherone was able to identify hundreds of distinct miRNAs, which — despite their sequence differences — bound many of the same mRNAs. Of all the tissues she examined, the brain appeared to have the most co-targeting. So she narrowed her focus to explore the overlapping functions of just two miRNAs that worked together there: miR-138 and miR-137.
“That was a really interesting observation and a functional demonstration of the overlap between these two miRNAs,” she says. “One miRNA can rescue the loss of a completely different miRNA if they share targets.”If she deleted miR-138 from her cells, they could no longer differentiate and become neurons. However, when she added miR-138’s co-targeting partner, miR-137, the cells were once again able to differentiate.
Cherone went on to identify an entire group of miRNAs within the brain, nine in total, that also shared similar targets. She selected several genes targeted by three or more of these miRNAs, and mutated every possible combination of the miRNA sites to determine their individual contributions. She ultimately established that subsets of the miRNAs could repress gene expression between five- and tenfold if they were expressed at the same time and bound close together.
According to Cherone, “seeing a tenfold repression by miRNAs is unheard of.” Such strong repression can have serious phenotypic consequences. She attributes this finding to the lab’s advanced computational strategies, which allowed them to systematically and unbiasedly identify the miRNAs that work together and their gene targets.
Why might a single gene be regulated by so many different miRNAs? There are more evolutionary paths to acquire sites for many different miRNAs than paths to acquire sites for the same miRNA. And, the authors explain, this arrangement may allow more precise control of cell type-specific expression.
Given that their miRNAs of interest primarily worked in the brain, the researchers wondered why this tissue might require so much co-targeting. One idea is that mRNAs in the brain tend to have longer regions where more miRNAs can bind to exert their effects. Another possibility is that mRNA expression in the brain must be especially fine-tuned, because too much or too little expression could have severe ramifications for neuronal function and development. For instance, fragile X-associated tremor/ataxia syndrome (FXTAS) can result from fairly subtle changes in proteins levels.
“Co-targeting appears to be widespread in many tissues, not just the brain,” says senior author Christopher Burge, a professor of biology at MIT. “This means that strategies to modulate the activity of a miRNA in a genetic or therapeutic context will be most effective when they take into account the levels of the other miRNAs that frequently partner with the miRNA of interest.”
“It’s time to start thinking of miRNAs as working together in networks, rather than functioning as individual units,” Cherone says. “If you want to know the function of a given miRNA, you have to understand the group it’s collaborating with, and explore its function within that group.”
Top image: Graphical illustration of co-targeting by miRNAs. Credit: Jennifer Cherone.
Citation:
“Cotargeting among microRNAs in the brain.”
Genome Research, online October 24, 2019, DOI: 10.1101/gr.249201.119
Jennifer M. Cherone, Vjola Jorgji, and Christopher B. Burge.