By unraveling the genetic pathways that help Toxoplasma gondii persist in human cells, Sebastian Lourido hopes to find new ways to treat toxoplasmosis.
Anne Trafton | MIT News
August 25, 2024
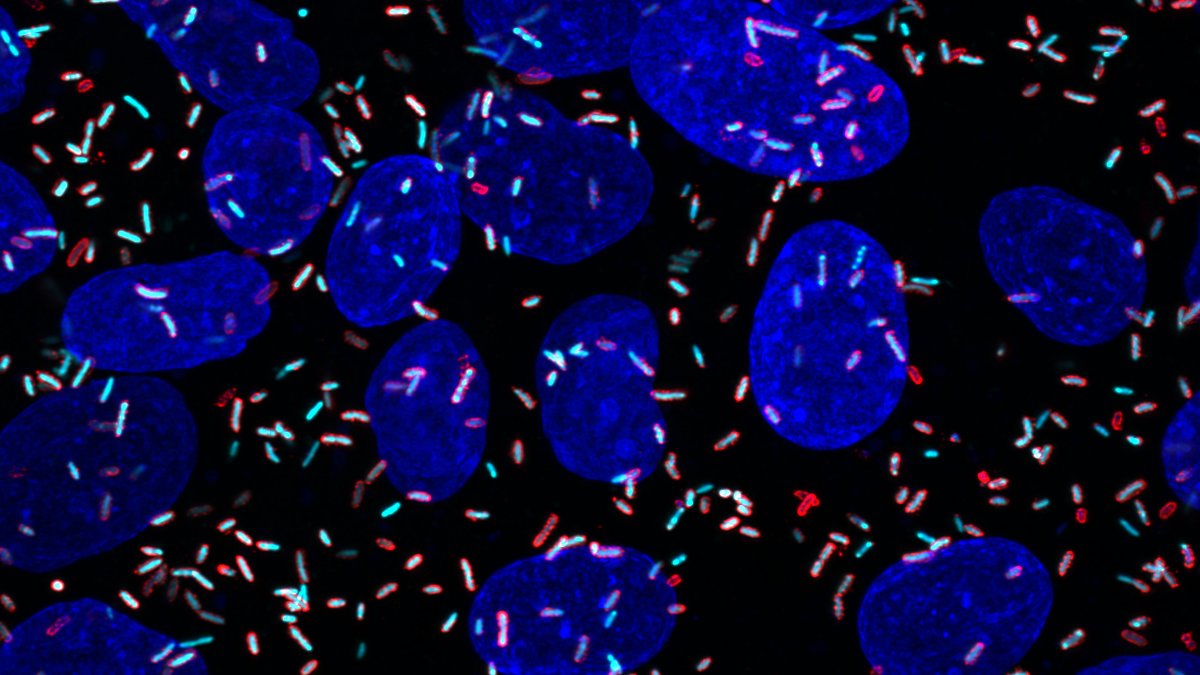
Toxoplasma gondii, the parasite that causes toxoplasmosis, is believed to infect as much as one-third of the world’s population. Many of those people have no symptoms, but the parasite can remain dormant for years and later reawaken to cause disease in anyone who becomes immunocompromised.
Why this single-celled parasite is so widespread, and what triggers it to reemerge, are questions that intrigue Sebastian Lourido, an associate professor of biology at MIT and member of the Whitehead Institute for Biomedical Research. In his lab, research is unraveling the genetic pathways that help to keep the parasite in a dormant state, and the factors that lead it to burst free from that state.
“One of the missions of my lab to improve our ability to manipulate the parasite genome, and to do that at a scale that allows us to ask questions about the functions of many genes, or even the entire genome, in a variety of contexts,” Lourido says.
There are drugs that can treat the acute symptoms of Toxoplasma infection, which include headache, fever, and inflammation of the heart and lungs. However, once the parasite enters the dormant stage, those drugs don’t affect it. Lourido hopes that his lab’s work will lead to potential new treatments for this stage, as well as drugs that could combat similar parasites such as a tickborne parasite known as Babesia, which is becoming more common in New England.
“There are a lot of people who are affected by these parasites, and parasitology often doesn’t get the attention that it deserves at the highest levels of research. It’s really important to bring the latest scientific advances, the latest tools, and the latest concepts to the field of parasitology,” Lourido says.
A fascination with microbiology
As a child in Cali, Colombia, Lourido was enthralled by what he could see through the microscopes at his mother’s medical genetics lab at the University of Valle del Cauca. His father ran the family’s farm and also worked in government, at one point serving as interim governor of the state.
“From my mom, I was exposed to the ideas of gene expression and the influence of genetics on biology, and I think that really sparked an early interest in understanding biology at a fundamental level,” Lourido says. “On the other hand, my dad was in agriculture, and so there were other influences there around how the environment shapes biology.”
Lourido decided to go to college in the United States, in part because at the time, in the early 2000s, Colombia was experiencing a surge in violence. He was also drawn to the idea of attending a liberal arts college, where he could study both science and art. He ended up going to Tulane University, where he double-majored in fine arts and cell and molecular biology.
As an artist, Lourido focused on printmaking and painting. One area he especially enjoyed was stone lithography, which involves etching images on large blocks of limestone with oil-based inks, treating the images with chemicals, and then transferring the images onto paper using a large press.
“I ended up doing a lot of printmaking, which I think attracted me because it felt like a mode of expression that leveraged different techniques and technical elements,” he says.
At the same time, he worked in a biology lab that studied Daphnia, tiny crustaceans found in fresh water that have helped scientists learn about how organisms can develop new traits in response to changes to their environment. As an undergraduate, he helped develop ways to use viruses to introduce new genes into Daphnia. By the time he graduated from Tulane, Lourido had decided to go into science rather than art.
“I had really fallen in love with lab science as an undergrad. I loved the freedom and the creativity that came from it, the ability to work in teams and to build on ideas, to not have to completely reinvent the entire system, but really be able to develop it over a longer period of time,” he says.
After graduating from college, Lourido spent two years in Germany, working at the Max Planck Institute for Infection Biology. In Arturo Zychlinksy’s lab, Lourido studied two bacteria known as Shigella and Salmonella, which can cause severe illnesses, including diarrhea. His studies there helped to reveal how these bacteria get into cells and how they modify the host cells’ own pathways to help them replicate inside cells.
As a graduate student at Washington University in St. Louis, Lourido worked in several labs focusing on different aspects of microbiology, including virology and bacteriology, but eventually ended up working with David Sibley, a prominent researcher specializing in Toxoplasma.
“I had not thought much about Toxoplasma before going to graduate school,” Lourido recalls. “I was pretty unaware of parasitology in general, despite some undergrad courses, which honestly very superficially treated the subject. What I liked about it was here was a system where we knew so little — organisms that are so different from the textbook models of eukaryotic cells.”
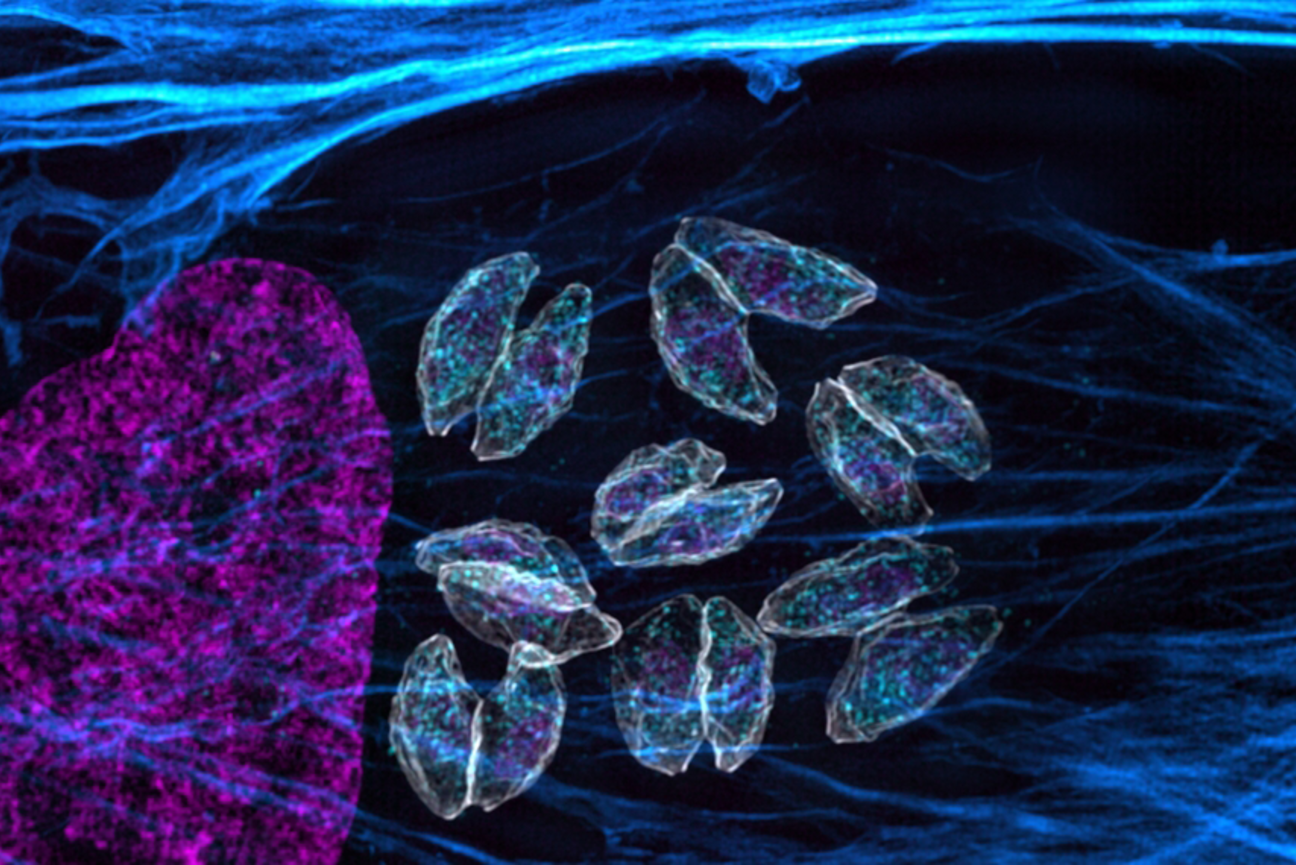
Toxoplasma gondii belongs to a group of parasites known as apicomplexans — a type of protozoans that can cause a variety of diseases. After infecting a human host, Toxoplasma gondii can hide from the immune system for decades, usually in cysts found in the brain or muscles. Lourido found the organism especially intriguing because as a 17-year-old, he had been diagnosed with toxoplasmosis. His only symptom was swollen glands, but doctors found that his blood contained antibodies against Toxoplasma.
“It is really fascinating that in all of these people, about a quarter to a third of the world’s population, the parasite persists. Chances are I still have live parasites somewhere in my body, and if I became immunocompromised, it would become a big problem. They would start replicating in an uncontrolled fashion,” he says.
A transformative approach
One of the challenges in studying Toxoplasma is that the organism’s genetics are very different from those of either bacteria or other eukaryotes such as yeast and mammals. That makes it harder to study parasitic gene functions by mutating or knocking out the genes.
Because of that difficulty, it took Lourido his entire graduate career to study the functions of just a couple of Toxoplasma genes. After finishing his PhD, he started his own lab as a fellow at the Whitehead Institute and began working on ways to study the Toxoplasma genome at a larger scale, using the CRISPR genome-editing technique.
With CRISPR, scientists can systematically knock out every gene in the genome and then study how each missing gene affects parasite function and survival.
“Through the adaptation of CRISPR to Toxoplasma, we’ve been able to survey the entire parasite genome. That has been transformative,” says Lourido, who became a Whitehead member and MIT faculty member in 2017. “Since its original application in 2016, we’ve been able to uncover mechanisms of drug resistance and susceptibility, trace metabolic pathways, and explore many other aspects of parasite biology.”
Using CRISPR-based screens, Lourido’s lab has identified a regulatory gene called BFD1 that appears to drive the expression of genes that the parasite needs for long-term survival within a host. His lab has also revealed many of the molecular steps required for the parasite to shift between active and dormant states.
“We’re actively working to understand how environmental inputs end up guiding the parasite in one direction or another,” Lourido says. “They seem to preferentially go into those chronic stages in certain cells like neurons or muscle cells, and they proliferate more exuberantly in the acute phase when nutrient conditions are appropriate or when there are low levels of immunity in the host.”