Greta Friar | Whitehead Institute
October 9, 2018
Cambridge, Mass. — Brain development is a delicately choreographed dance in which cell division and differentiation into mature cell types must be performed in the right balance for normal growth. In order to better understand factors affecting brain development, Whitehead Institute researchers investigated a genetic mutation that leads to a brain-specific developmental disorder in spite of the gene’s prevalent expression in other cell types.
Kinetochore null protein 1 (KNL1) acts throughout the body during cell division to help ensure the accurate segregation of chromosomes into each daughter cell. A mutation in the KNL1 gene caused by a single change in its DNA sequence leads to microcephaly, a condition in which the brain fails to properly develop, causing babies to be born with small heads, often accompanied by intellectual disabilities and other health problems. In an article published online October 9 in the journal Cell Reports, Whitehead Institute Founding Member Rudolf Jaenisch and colleagues investigated how this KNL1 mutation can lead to microcephaly without affecting other cell types, providing important insights into the underlying basis of microcephaly and the role that KNL1 normally plays in brain development.
“The key question we were interested in was why, if the gene is ubiquitously expressed, is there a brain-specific phenotype,” says Jaenisch, who is also a professor of biology at the Massachusetts Institute of Technology.
Jaenisch lab graduate student Attya Omer Javed, a co-first author on the paper along with past lab members Yun Li and Julien Muffat, used CRISPR-Cas9 to recreate the mutation—a point mutation, or one-letter change in the DNA sequence of the KNL1 gene—in several different cell types derived from human stem cells in the lab. Of the three cell types tested, they found that only the neural progenitor cells, early stage cells that become brain cells, appeared to be affected.
As the brain develops, each neural progenitor can either keep dividing to increase the overall number of cells in the brain, or it can mature into a differentiated brain cell, at which point it is no longer able to divide. For a healthy brain to develop, there needs to be a careful balance between these two processes of proliferation and differentiation. If the progenitors take too long to differentiate, the developing brain won’t have the specific cells it needs to assemble. But if all of the cells differentiate too quickly, before they can divide, there will be a shortage of cells and the brain will be too small.
“Neural progenitors are going through many cell cycles, dividing quickly during brain development. Even a small defect could accumulate to have a huge impact,” Omer Javed says.
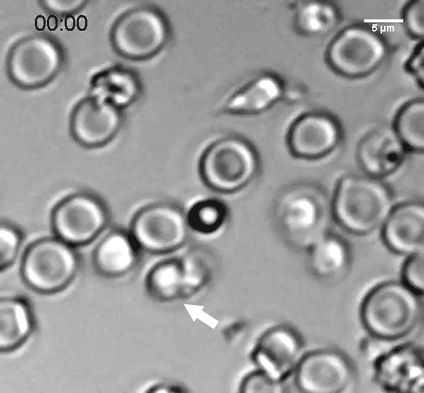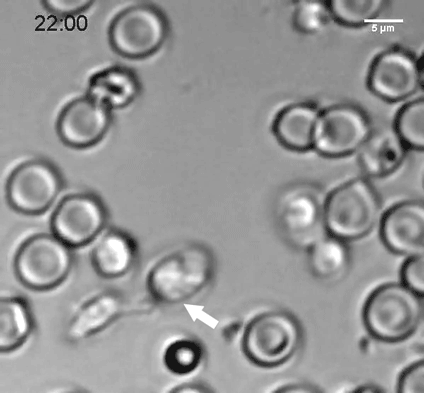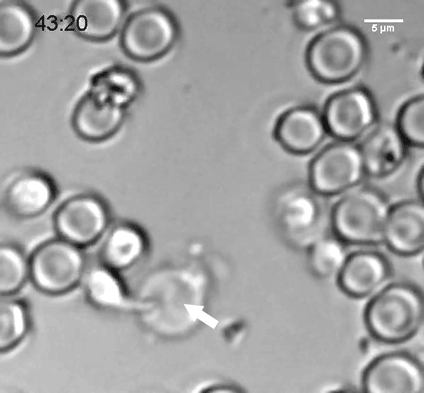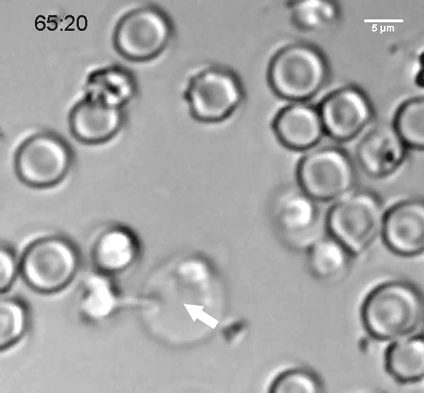
The researchers discovered that neural progenitors with the KNL1 mutation differentiated prematurely at the cost of proliferation, resulting in the small brain size that characterizes microcephaly. The brain cells with the mutation also were at a greater risk of cell death, disruption of the cell cycle, ending up with the wrong number of chromosomes, and malfunctions during attempted cell division.
KNL1’s role is in the kinetochore, an assembly of proteins that operate during mitosis to attach chromosomes to the machinery that will pull them apart into the daughter cells. This is why the KNL1 mutation negatively affects cell division. Co-author and Whitehead Member Iain Cheesemanhelped identify KNL1’s role in the kinetochore as a postdoctoral researcher years ago, and his expertise provided an opportunity for collaboration between his lab and Jaenisch’s.
“I have always found it interesting that inherited mutations to the kinetochore seem to lead to microcephaly,” Cheeseman says. “Investigating KNL1 together was an exciting chance to combine our labs’ diverse scientific knowledge.”
In order for the researchers to study the cells in an environment that more closely mimicked a human brain, they used a 3D cell culture technique to grow organoids made up of neural progenitors. Omer Javed found that the neural progenitors were extremely sensitive, as the organoids with the mutation expressed the microcephaly phenotype after as little as two weeks of growth.
Omer Javed then looked for differences between neural progenitors and the other cell types that would explain the brain-specific effects of the mutation. Even with the mutation, the KNL1 gene appeared able to make a functioning protein, explaining its lack of effect on the other cell types. So Omer Javed turned her attention to factors involved in regulating gene expression. For many of our genes to be expressed, first sections called introns must be removed, or spliced out, in order for the correct DNA sequence to then be read into RNA and then translated into a functional protein.
Omer Javed found that the KNL1 mutation created a site for splicing inhibitors to bind and silence the KNL1 gene by preventing it from being read into RNA. She also found a disparity in the level of a protein involved in this process between the cell types: the inhibitory splicing protein hnRNPA1 was much more prevalent in neural progenitors than elsewhere. When hnRNPA1 came across the site caused by the mutation, it prevented the gene from being expressed. The high quantity of hnRNPA1 in neural progenitors appears to be the main factor mediating the brain-specific effects of the mutation.
The work complements and extends previous investigations by the researchers into how neural progenitor proliferation may have contributed to the evolution of large human brains, as well as studies investigating why neural progenitors are so vulnerable to the Zika virus, which has been associated with microcephaly. Given their work suggesting that KNL1 could be a regulator of brain size, Omer Javed hopes that future research will reveal its role in the evolution of the human brain.
This research was supported by Boehringer Ingelheim Fonds, the Simons Foundation, the International Rett Syndrome Foundation, Brain & Behavior Research Foundation, the European Leukodystrophy Association, the National Institutes of Health (NIH grants HD 045022, R37-CA084198 and 1U19AI131135-01). Jaenisch is a cofounder of Fate Therapeutics, Fulcrum Therapeutics, and Omega Therapeutics.