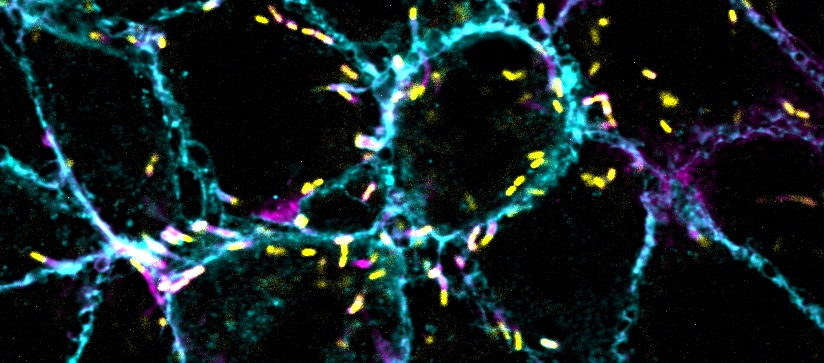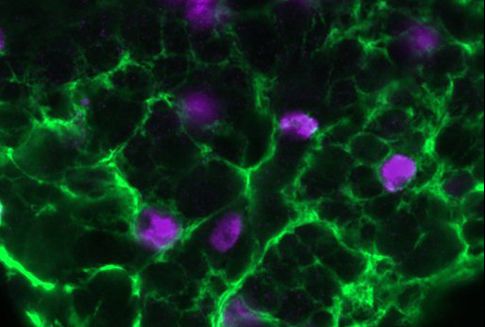
Molecules called ketone bodies may improve stem cells’ ability to regenerate new intestinal tissue.
Anne Trafton | MIT News Office
August 22, 2019
MIT biologists have discovered an unexpected effect of a ketogenic, or fat-rich, diet: They showed that high levels of ketone bodies, molecules produced by the breakdown of fat, help the intestine to maintain a large pool of adult stem cells, which are crucial for keeping the intestinal lining healthy.
The researchers also found that intestinal stem cells produce unusually high levels of ketone bodies even in the absence of a high-fat diet. These ketone bodies activate a well-known signaling pathway called Notch, which has previously been shown to help regulate stem cell differentiation.
“Ketone bodies are one of the first examples of how a metabolite instructs stem cell fate in the intestine,” says Omer Yilmaz, the Eisen and Chang Career Development Associate Professor of Biology and a member of MIT’s Koch Institute for Integrative Cancer Research. “These ketone bodies, which are normally thought to play a critical role in energy maintenance during times of nutritional stress, engage the Notch pathway to enhance stem cell function. Changes in ketone body levels in different nutritional states or diets enable stem cells to adapt to different physiologies.”
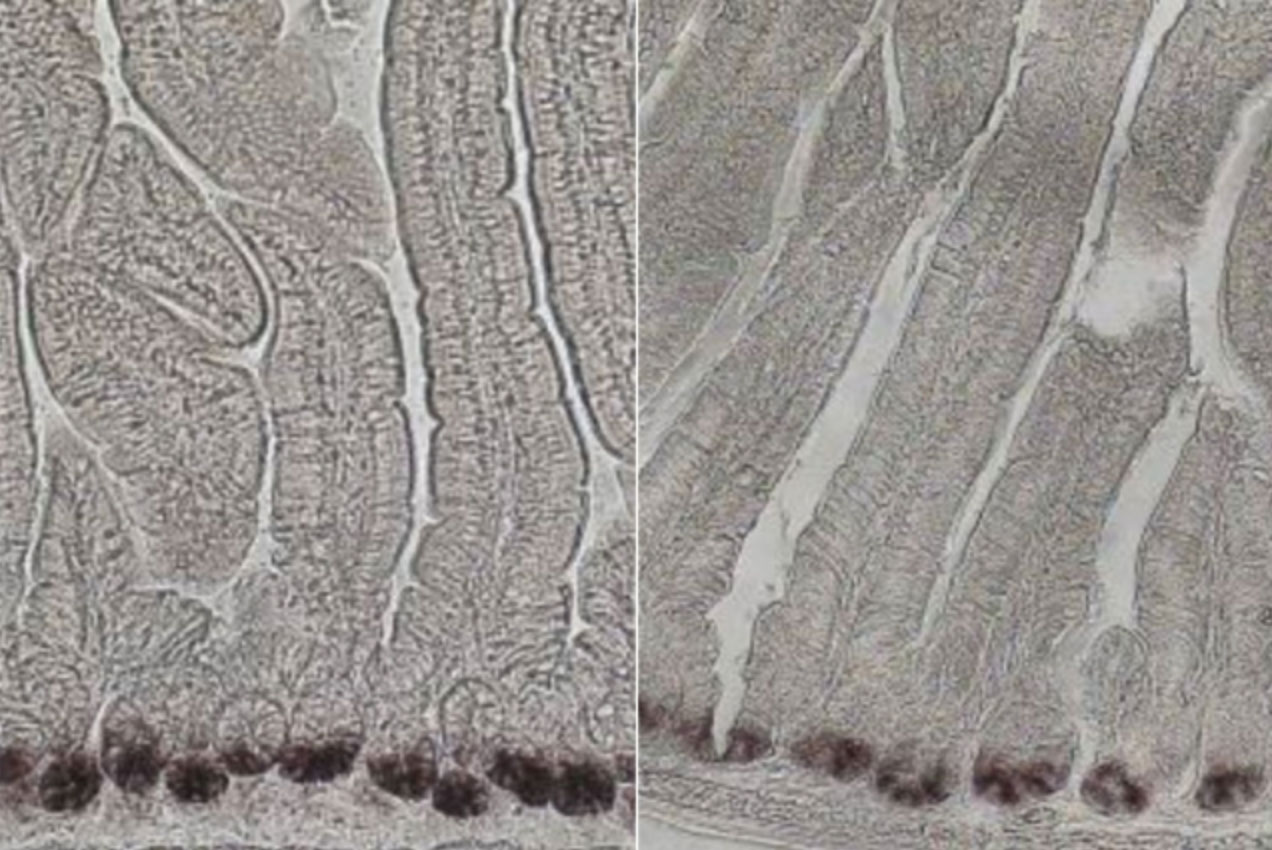
In a study of mice, the researchers found that a ketogenic diet gave intestinal stem cells a regenerative boost that made them better able to recover from damage to the intestinal lining, compared to the stem cells of mice on a regular diet.
Yilmaz is the senior author of the study, which appears in the Aug. 22 issue of Cell. MIT postdoc Chia-Wei Cheng is the paper’s lead author.
An unexpected role
Adult stem cells, which can differentiate into many different cell types, are found in tissues throughout the body. These stem cells are particularly important in the intestine because the intestinal lining is replaced every few days. Yilmaz’ lab has previously shown that fasting enhances stem cell function in aged mice, and that a high-fat diet can stimulate rapid growth of stem cell populations in the intestine.
In this study, the research team wanted to study the possible role of metabolism in the function of intestinal stem cells. By analyzing gene expression data, Cheng discovered that several enzymes involved in the production of ketone bodies are more abundant in intestinal stem cells than in other types of cells.
When a very high-fat diet is consumed, cells use these enzymes to break down fat into ketone bodies, which the body can use for fuel in the absence of carbohydrates. However, because these enzymes are so active in intestinal stem cells, these cells have unusually high ketone body levels even when a normal diet is consumed.
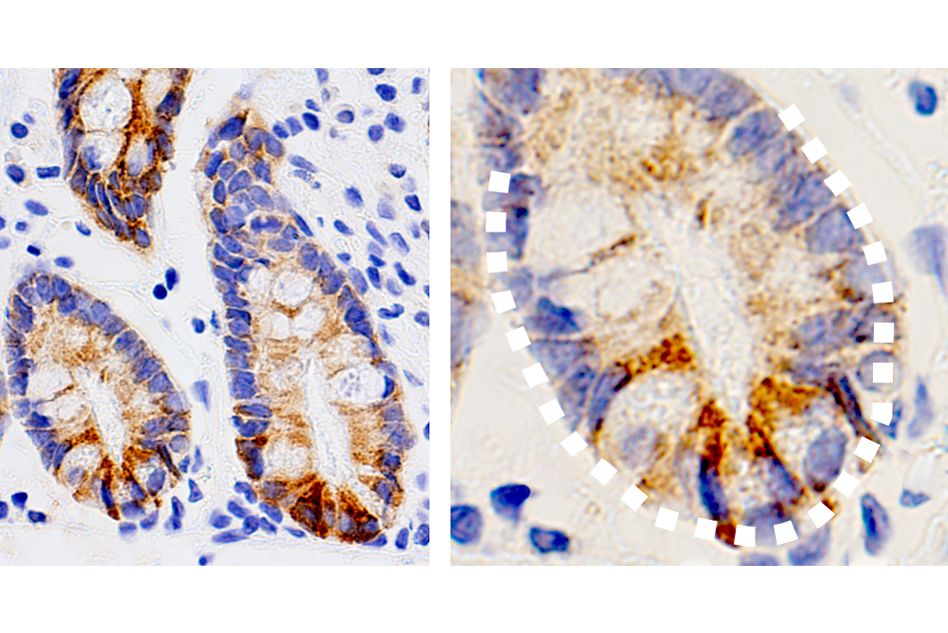
To their surprise, the researchers found that the ketones stimulate the Notch signaling pathway, which is known to be critical for regulating stem cell functions such as regenerating damaged tissue.
“Intestinal stem cells can generate ketone bodies by themselves, and use them to sustain their own stemness through fine-tuning a hardwired developmental pathway that controls cell lineage and fate,” Cheng says.
In mice, the researchers showed that a ketogenic diet enhanced this effect, and mice on such a diet were better able to regenerate new intestinal tissue. When the researchers fed the mice a high-sugar diet, they saw the opposite effect: Ketone production and stem cell function both declined.
Stem cell function
The study helps to answer some questions raised by Yilmaz’ previous work showing that both fasting and high-fat diets enhance intestinal stem cell function. The new findings suggest that stimulating ketogenesis through any kind of diet that limits carbohydrate intake helps promote stem cell proliferation.
“Ketone bodies become highly induced in the intestine during periods of food deprivation and play an important role in the process of preserving and enhancing stem cell activity,” Yilmaz says. “When food isn’t readily available, it might be that the intestine needs to preserve stem cell function so that when nutrients become replete, you have a pool of very active stem cells that can go on to repopulate the cells of the intestine.”
The findings suggest that a ketogenic diet, which would drive ketone body production in the intestine, might be helpful for repairing damage to the intestinal lining, which can occur in cancer patients receiving radiation or chemotherapy treatments, Yilmaz says.
The researchers now plan to study whether adult stem cells in other types of tissue use ketone bodies to regulate their function. Another key question is whether ketone-induced stem cell activity could be linked to cancer development, because there is evidence that some tumors in the intestines and other tissues arise from stem cells.
“If an intervention drives stem cell proliferation, a population of cells that serve as the origin of some tumors, could such an intervention possibly elevate cancer risk? That’s something we want to understand,” Yilmaz says. “What role do these ketone bodies play in the early steps of tumor formation, and can driving this pathway too much, either through diet or small molecule mimetics, impact cancer formation? We just don’t know the answer to those questions.”
The research was funded by the National Institutes of Health, a V Foundation V Scholar Award, a Sidney Kimmel Scholar Award, a Pew-Stewart Trust Scholar Award, the MIT Stem Cell Initiative, the Koch Institute Frontier Research Program through the Kathy and Curt Marble Cancer Research Fund, the Koch Institute Dana Farber/Harvard Cancer Center Bridge Project, and the American Federation of Aging Research.