The need to produce just the right amount of protein is behind the striking uniformity of sizes.
Anne Trafton | MIT News Office
February 7, 2019
MIT biologists have discovered the answer to a fundamental biological question: Why are cells of a given type all the same size?
In humans, cell size can vary more than 100-fold, ranging from tiny red blood cells to large neurons. However, within each cell type, there is very little deviation from a standard size. In studies of yeast, MIT researchers grew cells to 10 times their normal size and found that their DNA could not keep up with the demands of producing enough protein to maintain normal cell functions.
Furthermore, the researchers found that this protein shortage leads the cells into a nondividing state known as senescence, suggesting a possible explanation for how cells become senescent as they age.
“There are so many hypotheses out there that try to explain why senescence happens, and I think this data provides a beautiful and simple explanation for senescence,” says Angelika Amon, the Kathleen and Curtis Marble Professor in Cancer Research in the Department of Biology and a member of the Koch Institute for Integrative Cancer Research.
Amon is the senior author of the study, which appears in the Feb. 7 online edition of Cell. Gabriel Neurohr, an MIT postdoc, is the lead author of the paper.
Excessive size
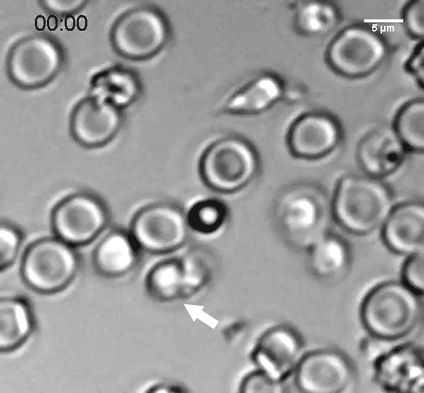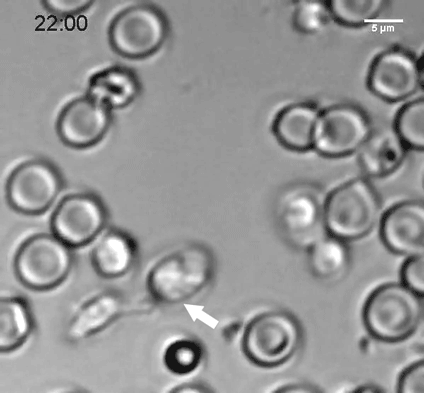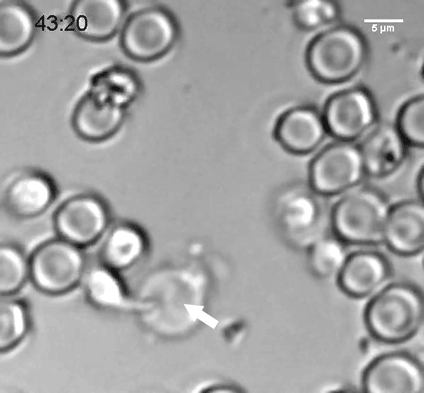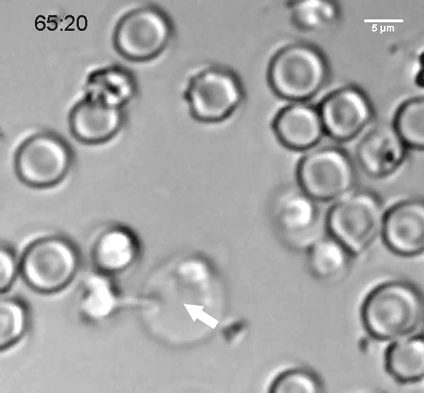
To explore why cell size is so tightly controlled, the researchers prevented yeast cells from dividing by modifying a gene critical for cell division, so that it could be turned off at a certain temperature. These cells continued to grow, but they could not divide and they did not replicate their DNA.
The researchers discovered that as the cells expanded, their DNA and their protein-building machinery could not keep pace with the needs of such a large cell. This failure to produce enough protein led to the dilution of the cytoplasm and disruption of cell division. The researchers believe that many other fundamental cell processes that rely on cellular molecules finding and interacting with each other may also be impaired when cells are too big.
“Theoretical models predict that diluting the cytoplasm will decrease reaction rates. Every chemical reaction would occur more slowly, and some threshold concentrations of certain proteins may not be reached, so certain reactions would never happen because the concentrations are lower,” Neurohr says.
The researchers showed that yeast cells with two sets of chromosomes were able to grow to twice the size of yeast cells with just one set of chromosomes before becoming senescent, suggesting that the amount of DNA in the cells is the limiting factor in the cells’ ability to grow.
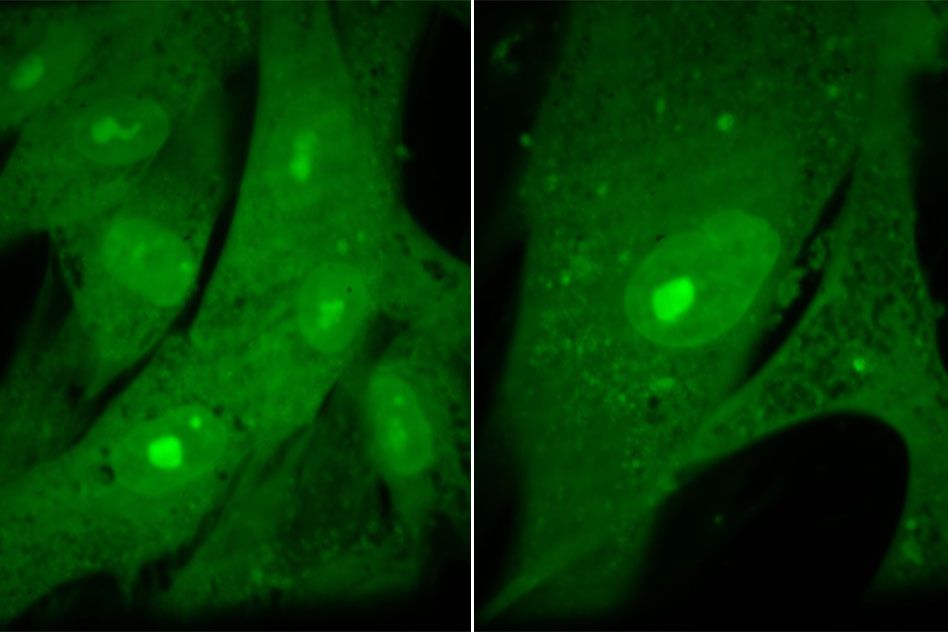
Experiments with human cells yielded similar results: In a study of human fibroblast cells, the researchers found that forcing the cells to grow to excessive sizes (eight times their normal size) disrupted many functions, including cell division.
“It’s been clear for some time that cells do control their size, but it’s been unclear what the various physiological reasons are for why they do so,” says Jan Skotheim, an associate professor of biology at Stanford University, who was not involved in the research. “What’s nice about this work is it really shows how things go wrong when cells get too big.”
Age-related disease
Amon says excessive growth likely plays a major role in the development of senescence, which occurs in many types of mammalian cells and is thought to contribute to age-related organ dysfunction and chronic age-related diseases.
Senescent cells are often larger than younger cells, and this study, which showed that unchecked cell growth leads to senescence, offers a possible explanation for this observation. Human cells tend to grow slightly larger throughout their lifetimes, because every time a cell divides, it checks for DNA damage, and if any is found, division is halted while repairs are made. During each of these delays, the cell grows slightly larger.
“Over the lifetime of a cell, the more divisions you make, the higher your probability is of having that damage, and over time cells will get larger,” Amon says. “Eventually they get so large that they start diluting critical factors that are important for proliferation.”
A difficult question that remains unanswered is how different types of cells maintain the appropriate size for their cell type, which the researchers now hope to study further.
The research was funded, in part, by the National Institutes of Health, the Howard Hughes Medical Institute, the Paul F. Glenn Center for Biology of Aging Research at MIT, a National Science Foundation graduate research fellowship, the William Bowes Fellows program, and the Vilcek Foundation.