In the lab, Biology Professor Amy Keating researches the interactions of proteins with a mix of modeling and synthetic lab work and diverse minds
School of Science
June 11, 2020
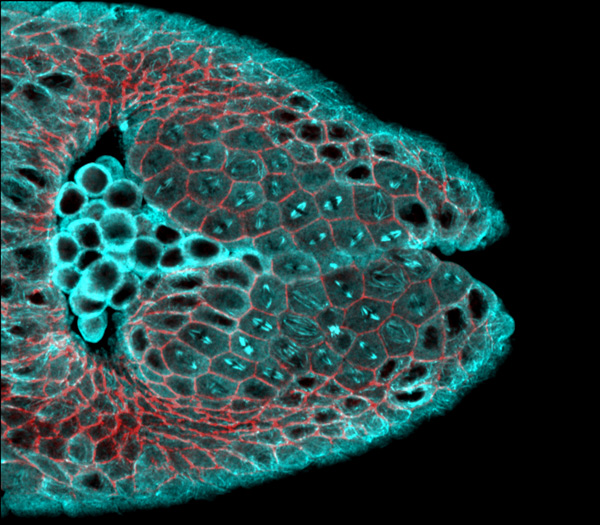
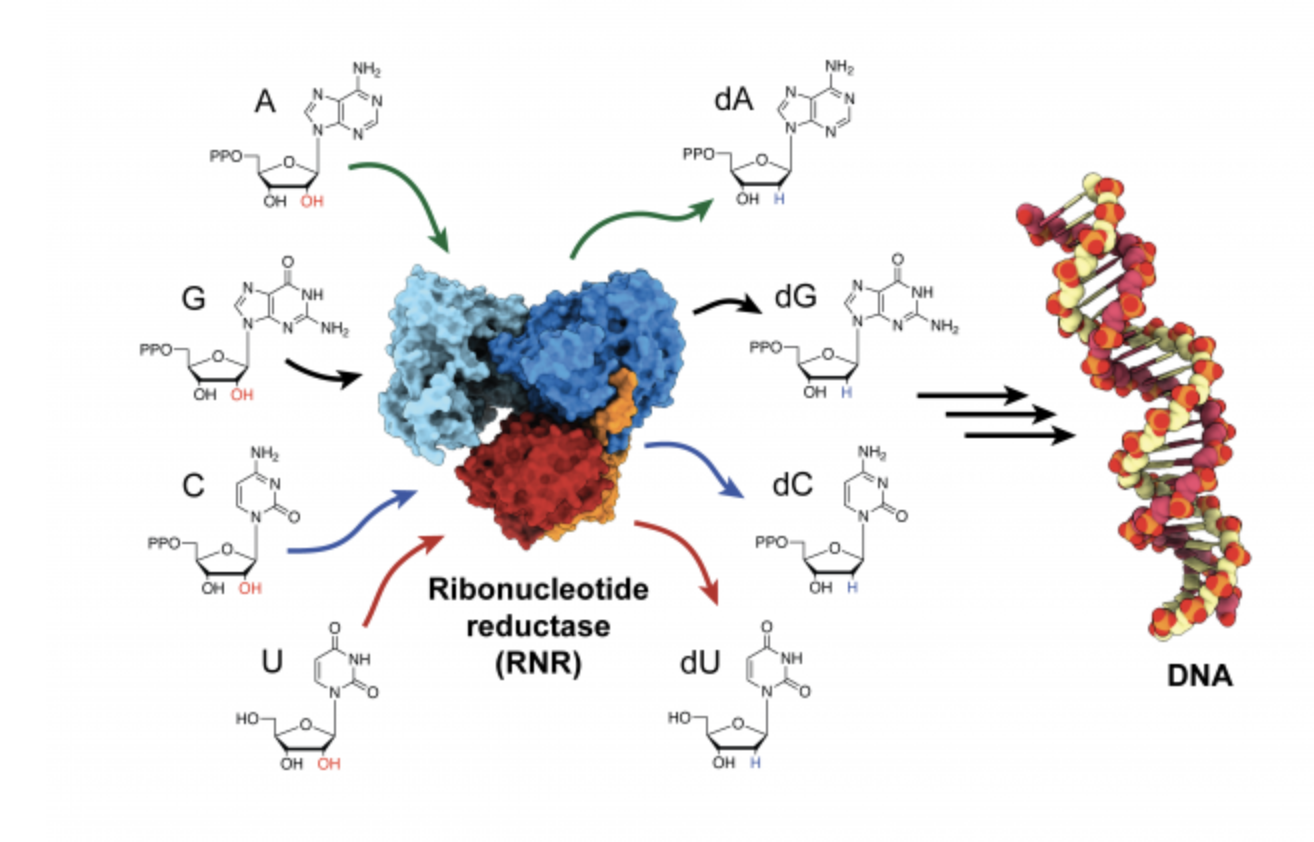
Almost everything in biology is a multistep process, from the metabolization of carbohydrates and fats as fuel to information transcription from DNA and RNA. Without proteins and their interactions, cells couldn’t perform any of these biological tasks. But how do proteins establish their individual roles? And how do they interact with each other? These questions drive Professor Amy Keating’s research, and both lab experiments and computational modeling are helping her reveal the mysteries behind the basic functions of life.
In Keating’s field of research, as with most areas of science, the use of artificial intelligence is a relatively new – and growing – trend. “It’s pretty scary how fast new methods in machine learning are changing the landscape,” says Keating, who holds appointments in both the Department of Biology and the Department of Biological Engineering. “I think that we will see a disruptive change in protein modeling over the coming years.” She has found that incorporating basic machine learning methods in her own work has generated some success in uncovering how protein sequences determine their interactions.
However, there are limits to using only computational modeling due to the complexities of protein-protein interaction and a general need for empirical data to calibrate the models. Her lab group integrates computation with biological engineering in a laboratory setting. Keating’s team often starts by using computational modeling to narrow down their search from a massive collection of protein structure models. This step limits their output from an effectively infinite space (~1030) to something on the order of 106 potential promising molecules that can be experimentally tested. They can feed the results of experiments into other algorithms that help designate the specific features of the protein that prove important. This process is cyclical, and Keating emphasizes that experimental efforts are crucial for improving the success rate of this kind of work. That is where the lab comes in. There, they do what the computer cannot: they build proteins.
With the disruption of the COVID-19 crisis the Keating lab has focused their attention on computational projects, as well as on reviewing the literature and writing up papers and theses. The members are also using their time at home to brainstorm and plan their research. “We are having multiple group meetings per week by Zoom, including a ‘Keating Group Idea Lab,’ at which everyone throws out ideas, ranging from practical suggestions about current projects to out-there new concepts, for group discussion,” says Keating. “We are confident that we can use this time productively, to advance our science, even as we make long lists of things that we are eager to do as soon as we can get back into the laboratory.”
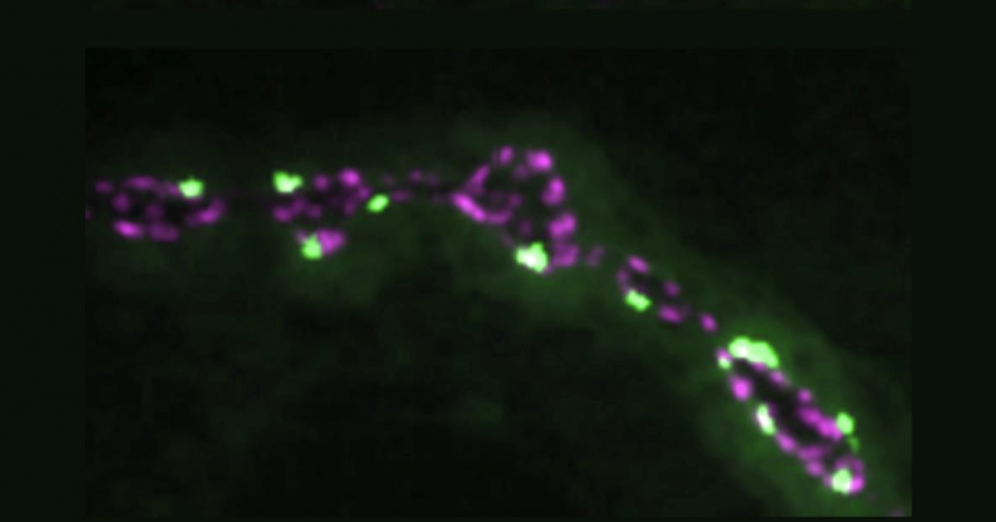
A topic of current interest to Keating and her group members is interactions among proteins with “short linear motifs” or SLiMs, which are abundant –more than one hundred thousand such motifs are thought to exist in one human. One family of these SLiM-binding proteins regulates movement of cells within the body and is implicated in the spread of cancer cells to a secondary location (metastasis). The lab’s novel mini-protein and peptide designs aim to disrupt these protein interactions and could be useful for eventually disrupting and treating cancer and other diseases.
FOSTERING MULTIPLE INTERACTIONS
Currently, Keating’s research team consists of six students who have backgrounds in almost as many different cultures. Her students’ diversity, which stems not just from different focuses in formal training but also from life experiences, is integral to their success, according to Keating. She wishes that more women like herself and members of underrepresented minority groups who love STEM would consider pursuing academic careers. “It’s hard work, but it’s very rewarding,” she entices. The best thing about being a faculty member, she believes, is having a team of bright minds who contribute unique ideas and insights to a problem and provide information beyond her own areas of expertise.
“I learn facts that they know and I do not. I learn interesting ways of thinking about science and also ways of doing science,” she says, noting that novel ideas in methodology lead to advances in research. “I’ve learned a lot of things about computer science from my students. I’m happy that one of my former biology students is [now] a professor of computer science,” she admits, appreciating his expertise as a benefit in frequent collaborations. “I love that students at MIT question everything.” Keating’s ever-expanding knowledge builds on top of a diverse background gleaned during her time as a student.
Keating’s bachelor’s degree from Harvard University is in physics. During her PhD at University of California, Los Angeles, she shifted to chemistry — specifically computational physical organic chemistry. When browsing for a postdoctoral position, she discovered the work of former MIT Department of Biology faculty and Whitehead Institute member Peter Kim and joined him. She maintained her interest in computation as a tool for biological research, concurrently co-advised by MIT Professor of Electrical Engineering and Computer Science Bruce Tidor. It was somewhat down to chance that her academic job search led her to MIT. “I certainly never thought I would be a biology professor, especially at MIT,” she remarks of her convoluted career path through the wide world of science.
But it is an unexpected result for which Keating is grateful. “My undergrad self would have been surprised by the MIT School of Science,” she muses, which makes MIT “so much more than ‘just’ the world’s best engineering school.” That is something of a common misconception about the Institute, she feels. “I think a lot of people outside of MIT don’t know how outstanding our basic science programs are.” Keating is a part of the strong science education at MIT, which is constantly adapting to keep up with the digital age, which led to her receiving the most recent Fund for the Future of Science Award.
“I was thrilled, and pretty surprised, to receive the award; my fantastic colleagues in the School of Science are not people that you want to be competing with.” This support is invaluable to her research on the foundations of biological interactions, and to ensure a robust team that has what it needs to develop important advances. The curious minds with which she collaborates are equally as invaluable.
“The people at MIT are amazingly smart, curious, and focused on things that I value,” Keating adds, “like good ideas, intellectual rigor, discovering new things, and education.”
This article appeared in the Summer 2020 issue of Science@MIT