Research Area: Cell Biology
The Davis and Berger labs combined cryo-electron microscopy and machine learning to visualize molecules in 3D.
February 4, 2021
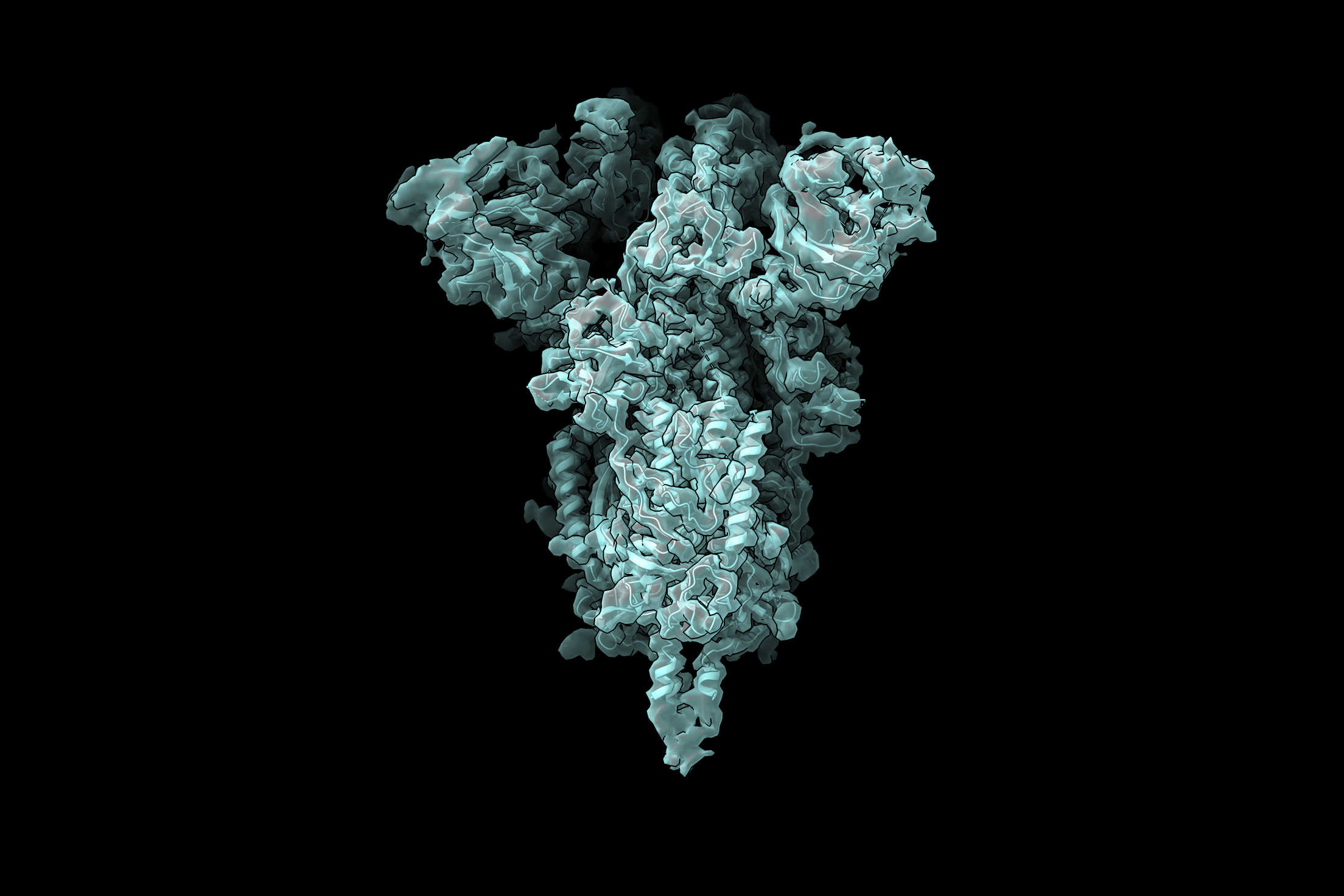
New technique reveals many possible conformations that a protein may take.
Anne Trafton | MIT News Office
February 4, 2021
Cryo-electron microscopy (cryo-EM) allows scientists to produce high-resolution, three-dimensional images of tiny molecules such as proteins. This technique works best for imaging proteins that exist in only one conformation, but MIT researchers have now developed a machine-learning algorithm that helps them identify multiple possible structures that a protein can take.
Unlike AI techniques that aim to predict protein structure from sequence data alone, protein structure can also be experimentally determined using cryo-EM, which produces hundreds of thousands, or even millions, of two-dimensional images of protein samples frozen in a thin layer of ice. Computer algorithms then piece together these images, taken from different angles, into a three-dimensional representation of the protein in a process termed reconstruction.
In a Nature Methods paper, the MIT researchers report a new AI-based software for reconstructing multiple structures and motions of the imaged protein — a major goal in the protein science community. Instead of using the traditional representation of protein structure as electron-scattering intensities on a 3D lattice, which is impractical for modeling multiple structures, the researchers introduced a new neural network architecture that can efficiently generate the full ensemble of structures in a single model.
“With the broad representation power of neural networks, we can extract structural information from noisy images and visualize detailed movements of macromolecular machines,” says Ellen Zhong, an MIT graduate student and the lead author of the paper.
With their software, they discovered protein motions from imaging datasets where only a single static 3D structure was originally identified. They also visualized large-scale flexible motions of the spliceosome — a protein complex that coordinates the splicing of the protein coding sequences of transcribed RNA.
“Our idea was to try to use machine-learning techniques to better capture the underlying structural heterogeneity, and to allow us to inspect the variety of structural states that are present in a sample,” says Joseph Davis, the Whitehead Career Development Assistant Professor in MIT’s Department of Biology.
Davis and Bonnie Berger, the Simons Professor of Mathematics at MIT and head of the Computation and Biology group at the Computer Science and Artificial Intelligence Laboratory, are the senior authors of the study, which appears today in Nature Methods. MIT postdoc Tristan Bepler is also an author of the paper.
Visualizing a multistep process
The researchers demonstrated the utility of their new approach by analyzing structures that form during the process of assembling ribosomes — the cell organelles responsible for reading messenger RNA and translating it into proteins. Davis began studying the structure of ribosomes while a postdoc at the Scripps Research Institute. Ribosomes have two major subunits, each of which contains many individual proteins that are assembled in a multistep process.
To study the steps of ribosome assembly in detail, Davis stalled the process at different points and then took electron microscope images of the resulting structures. At some points, blocking assembly resulted in accumulation of just a single structure, suggesting that there is only one way for that step to occur. However, blocking other points resulted in many different structures, suggesting that the assembly could occur in a variety of ways.
Because some of these experiments generated so many different protein structures, traditional cryo-EM reconstruction tools did not work well to determine what those structures were.
“In general, it’s an extremely challenging problem to try to figure out how many states you have when you have a mixture of particles,” Davis says.
After starting his lab at MIT in 2017, he teamed up with Berger to use machine learning to develop a model that can use the two-dimensional images produced by cryo-EM to generate all of the three-dimensional structures found in the original sample.
In the new Nature Methods study, the researchers demonstrated the power of the technique by using it to identify a new ribosomal state that hadn’t been seen before. Previous studies had suggested that as a ribosome is assembled, large structural elements, which are akin to the foundation for a building, form first. Only after this foundation is formed are the “active sites” of the ribosome, which read messenger RNA and synthesize proteins, added to the structure.
In the new study, however, the researchers found that in a very small subset of ribosomes, about 1 percent, a structure that is normally added at the end actually appears before assembly of the foundation. To account for that, Davis hypothesizes that it might be too energetically expensive for cells to ensure that every single ribosome is assembled in the correct order.
“The cells are likely evolved to find a balance between what they can tolerate, which is maybe a small percentage of these types of potentially deleterious structures, and what it would cost to completely remove them from the assembly pathway,” he says.
Viral proteins
The researchers are now using this technique to study the coronavirus spike protein, which is the viral protein that binds to receptors on human cells and allows them to enter cells. The receptor binding domain (RBD) of the spike protein has three subunits, each of which can point either up or down.
“For me, watching the pandemic unfold over the past year has emphasized how important front-line antiviral drugs will be in battling similar viruses, which are likely to emerge in the future. As we start to think about how one might develop small molecule compounds to force all of the RBDs into the ‘down’ state so that they can’t interact with human cells, understanding exactly what the ‘up’ state looks like and how much conformational flexibility there is will be informative for drug design. We hope our new technique can reveal these sorts of structural details,” Davis says.
The research was funded by the National Science Foundation Graduate Research Fellowship Program, the National Institutes of Health, and the MIT Jameel Clinic for Machine Learning and Health. This work was supported by MIT Satori computation cluster hosted at the MGHPCC.
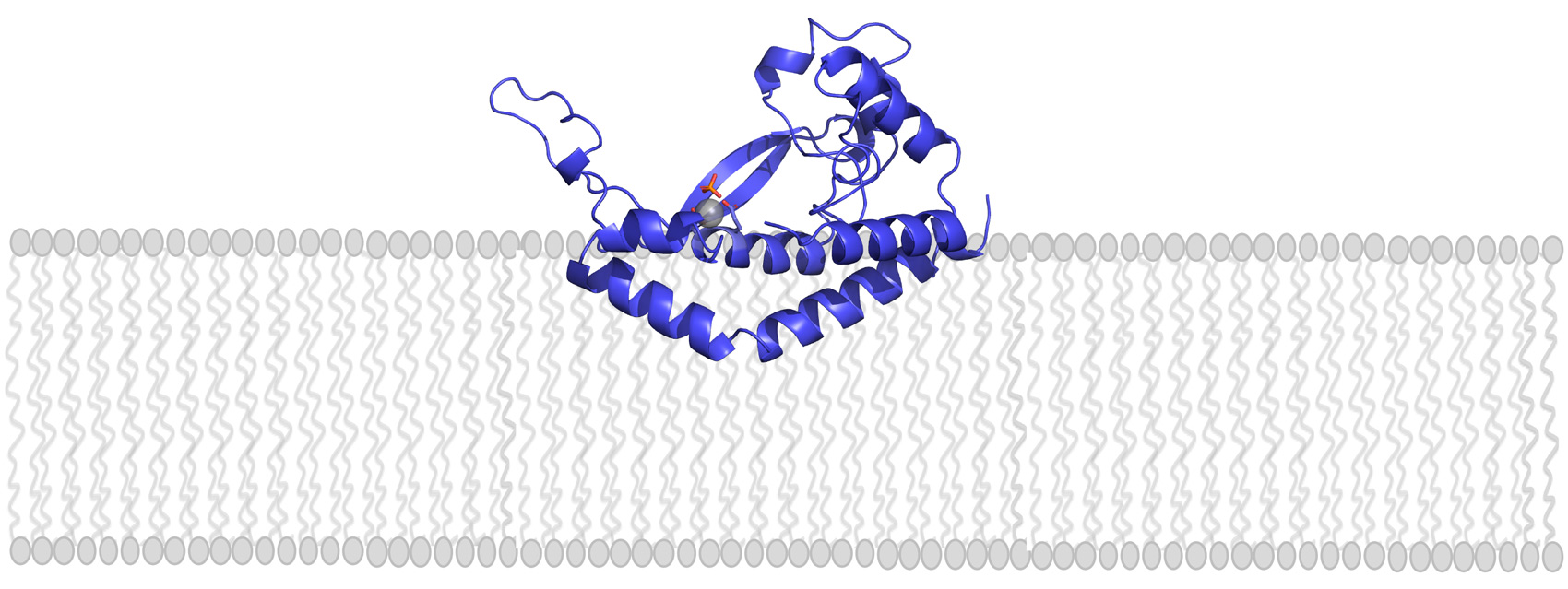
Raleigh McElvery
January 20, 2021
Many cells, including bacteria, are covered in a sugar-rich coating that protects their membrane and internal components. These sugars are often joined to other macromolecules, like proteins or lipids, to form glycoconjugates. The glycoconjugates that encrust bacteria help prevent them from “popping” under environmental stress, and facilitate host-pathogen interactions. Because the sugary layer perpetuates survival and virulence, researchers are looking for ways to create chinks in this microbial armor — or better yet, to prevent it from being made in the first place.
Glycoconjugates are built by many enzymes working in close succession at the cell membrane. One enzyme family, comprised of phosphoglycosyl transferases (PGTs), is responsible for catalyzing the first step in the assembly line. Of this large enzyme family, one subtype in particular stands out: “monotopic” PGTs, which are unique to bacteria and could serve as antibiotic targets. If researchers can develop drugs that inhibit monoPGTs, the sugar armor wouldn’t be built and noxious bacteria could be easier to defeat.
A new PNAS study co-authored by Professor of Biology and Chemistry, Barbara Imperiali, highlights the diversity and significance of these potential drug targets. Imperiali teamed up with graduate student Katherine O’Toole and Professor of Chemistry Karen Allen from Boston University to categorize over 38,000 different monoPGTs, compiling this information into the first database of its kind.
“We’ve taken an enzyme family that was once considered quirky and insignificant, and demonstrated that it’s actually very prevalent,” Imperiali says. “Hopefully the database will help us better understand these enzymes, their molecular pathways, and the human pathogens they support.”
Imperiali and her colleagues used sequence analysis of known monoPGTs to define a “signature” amino acid sequence. They leveraged this signature to identify the entire superfamily of monoPGTs amidst the 63,152 sequences downloaded from an online portal, which they then clustered into closely-related subtypes. The researchers also created a family tree, which included over 100 monoPGTs from diverse bacterial species. Imperiali hopes others will take advantage of this new information to pinpoint monoPGTs in pathogens of interest, and explore similarities and differences in related microbes and their enzymes.
The researchers’ analyses also revealed strange, new proteins that appeared to include two enzymes in one — a monoPGT fused to one of the other enzymes that typically play a separate role in the same sugar-modifying pathway. “It’s essentially one protein with two functions,” Imperiali explains. These fusion enzymes could reveal which enzymes “talk” to one another and work sequentially during the glycoconjugate-building process, she adds, revealing the complicated chain of events that creates the bacterial sugar shield.
The team even found cases where one monoPGT was fused to a member of a different PGT family — polytopic PGTs (polyPGTs). MonoPGTs and polyPGTs are involved in different pathways that each build glycoconjugates, so having a dual-function protein could allow cells to easily switch between mechanisms. Bacterial cells lack the organizational compartments that human and other eukaryotic cells have, so perhaps these fusion enzymes help exert control and order at different points in the cell cycle, Imperiali speculates. At the moment, though, the hybrid PGTs remain an evolutionary mystery.
While some researchers parse these ancient puzzles, others may use the database to inspire new drugs to combat antibiotic resistance. “At the end of the day,” Imperiali says, “we’ve shed light on a set of enzymes that could become pivotal therapeutic targets.”

Eva Frederick | Whitehead Institute
December 16, 2020
At any given moment in the human body, in about 30 trillion cells, DNA is being “read” into molecules of messenger RNA, the intermediary step between DNA and proteins, in a process called transcription.
Scientists have a pretty good idea of how transcription gets started: proteins called RNA polymerases are recruited to specific regions of the DNA molecules and begin skimming their way down the strand, synthesizing mRNA molecules as they go. But part of this process is less well understood: how does the cell know when to stop transcribing?
Now, new work from the labs of Whitehead Institute Member Richard Young, also a professor of biology at Massachusetts Institute of Technology (MIT), and Arup K. Chakraborty, professor of chemical engineering, physics and chemistry at MIT, suggests that RNA molecules themselves are responsible for regulating their formation through a feedback loop. Too few RNA molecules, and the cell initiates transcription to create more. Then, at a certain threshold, too many RNA molecules cause transcription to draw to a halt.
The research, published in Cell on December 16, represents a collaboration between biologists and physicists, and provides some insight into the potential roles of the thousands of RNAs that are not translated into any proteins, called noncoding RNAs, which are common in mammals and have mystified scientists for decades.
A question of condensates
Previous work in Young’s lab has focused on transcriptional condensates, small cellular droplets that bring together the molecules needed to transcribe DNA to RNA. Scientists in the lab discovered the transcriptional droplets in 2018, noticing that they typically formed when transcription began and dissolved a few seconds or minutes later when the process was finished.
The researchers wondered if the force that governed the dissolution of the transcriptional condensates could be related to the chemical properties of the RNA they produced — specifically, its highly negative charge. If this were the case, it would be the latest example of cellular processes being regulated via a feedback mechanism — an elegant, efficient system used in the cell to control biological functions such as red blood cell production and DNA repair.
As an initial test, the researchers used an in vitro experiment to test whether the amount of RNA had an effect on condensate formation. They found that within the range of physiological levels observed in cells, low levels of RNA encouraged droplet formation and high levels of RNA discouraged it.
Thinking outside the biology box
With these results in mind, Young Lab postdocs and co-first authors Ozgur Oksuz and Jon Henninger teamed up with physicist and co-first author Krishna Shrinivas, a graduate student in Arup Chakraborty’s lab, to investigate what physical forces were at play.
Shrinivas proposed that the team build a computational model to study the physical and chemical interactions between actively transcribed RNA and condensates formed by transcriptional proteins. The goal of the model was not to simply reproduce existing results, but to create a platform with which to test a variety of situations.
“The way most people study these kinds of problems is to take mixtures of molecules in a test tube, shake it and see what happens,” Shrinivas said. “That is as far away from what happens in a cell as one can imagine. Our thought was, ‘Can we try to study this problem in its biological context, which is this out-of-equilibrium, complex process?’”
Studying the problem from a physics perspective allowed the researchers to take a step back from traditional biology methods. “As a biologist, it’s difficult to come up with new hypotheses, new approaches to understanding how things work from available data,” Henninger said. “You can do screens, you can identify new players, new proteins, new RNAs that may be involved in a process, but you’re still limited by our classical understanding of how all these things interact. Whereas when talking with a physicist, you’re in this theoretical space extending beyond what the data can currently give you. Physicists love to think about how something would behave, given certain parameters.”
Once the model was complete, the researchers could ask it questions about situations that may arise in cells — for instance, what happens to condensates when RNAs of different lengths are produced at different rates as time ensues? — and then follow it up with an experiment at the lab bench. “We ended up with a very nice convergence of model and experiment,” Henninger said. “To me, it’s like the model helps distill the simplest features of this type of system, and then you can do more predictive experiments in cells to see if it fits that model.”
The charge is in charge
Through a series of modeling and experiments at the lab bench, the researchers were able to confirm their hypothesis that the effect of RNA on transcription is due to RNAs molecules’ highly negative charge. Furthermore, it was predicted that initial low levels of RNA enhance and subsequent higher levels dissolve condensates formed by transcriptional proteins. Because the charge is carried by the RNAs’ phosphate backbone, the effective charge of a given RNA molecule is directly proportional to its length.
In order to test this finding in a living cell, the researchers engineered mouse embryonic stem cells to have glowing condensates, then treated them with a chemical to disrupt the elongation phase of transcription. Consistent with the model’s predictions, the resulting dearth of condensate-dissolving RNA molecules increased the size and lifetime of condensates in the cell. Conversely, when the researchers engineered cells to induce the production of extra RNAs, transcriptional condensates at these sites dissolved. “These results highlight the importance of understanding how non-equilibrium feedback mechanisms regulate the functions of the biomolecular condensates present in cells,” said Chakraborty.
Confirmation of this feedback mechanism might help answer a long-standing mystery of the mammalian genome: the purpose of non-coding RNAs, which make up a large portion of genetic material. “While we know a lot about how proteins work, there are tens of thousands of noncoding RNA species, and we don’t know the functions of most of these molecules,” said Young. “The finding that RNA molecules can regulate transcriptional condensates makes us wonder if many of the noncoding species just function locally to tune gene expression throughout the genome. Then this giant mystery of what all these RNAs do has a potential solution.”
The researchers are optimistic that understanding this new role for RNA in the cell could inform therapies for a wide range of diseases. “Some diseases are actually caused by increased or decreased expression of a single gene,” said Oksuz, a co-first author. “We now know that if you modulate the levels of RNA, you have a predictable effect on condensates. So you could hypothetically tune up or down the expression of a disease gene to restore the expression — and possibly restore the phenotype — that you want, in order to treat a disease.”
Young added that a deeper understanding of RNA behavior could inform therapeutics more generally. In the last 10 years, a variety of drugs have been developed that directly target RNA successfully. “RNA is an important target,” Young said. “Understanding mechanistically how RNA molecules regulate gene expression bridges the gap between gene dysregulation in disease and new therapeutic approaches that target RNA.”

Greta Friar | Whitehead Institute
November 18, 2020
Melanosomes are the organelles, or structures, inside our cells, that produce melanin, the molecule that gives our skin, hair and eyes their color. Melanosomes produce several different forms of melanin, including black/brown coloration and yellow/red coloration, and the many variations in levels at which each coloration can be produced in an individual generate the wide variety of skin, hair, and eye colors in the world.
Many genes that have been associated with skin color encode proteins that are active in melanosomes, but their specific functions are unknown, leaving gaps in researchers’ understanding of the underlying biology of skin color. In order to help researchers get a more detailed understanding of melanosome biology, Whitehead Institute Member David Sabatini’s lab has developed a tool, called MelanoIP, with which researchers can rapidly and specifically isolate melanosomes from the cell and analyze their contents. Using this tool, researchers can uncover the identity of the proteins at work there and explain mechanistically how genetic variation contributes to differences in skin color. In research published in Nature on November 18, Sabatini and graduate student Charles Hank Adelmann unveil MelanoIP and explain how they used it to crack the identity of melanosome protein MFSD12.
MelanoIP is the latest in a series of tools based on a method that Sabatini, who is also a professor of biology at Massachusetts Institute of Technology and an investigator with the Howard Hughes Medical Institute, and collaborators developed to rapidly extract specific organelles from the cell for investigation. Sabatini and former graduate student Walter Chen first developed the method to isolate mitochondria. The process starts with researchers creating a tag that localizes to the organelle type of interest. Then they expose the contents of the whole cell to beads covered in antibodies that latch onto the tags, which pull the organelles with them when they are collected. The lab has since adapted this process to use on lysosomes, the recycling centers of the cell, and peroxisomes, organelles important in several metabolic processes—and now, melanosomes.
The first melanosome protein that Sabatini and Adelmann turned their attention to, MFSD12, was known to be linked to the production of red coloration or pheomelanin. When MFSD12 is suppressed, this leads to darker skin color in humans and mice, because the melanosomes are generating brown/black melanin but not any of the lighter red melanin. However, MFSD12’s exact role was unknown. Using MelanoIP, Adelmann discovered that MFSD12 is required for the import of the amino acid cysteine into melanosomes, which is a necessary component in red melanin synthesis. Adelmann’s research suggests that MFSD12 is itself the transporter, but further work is needed to confirm whether it works alone or in conjunction with other molecules.
One reason that the Sabatini lab picked the melanosome as the next organelle to apply their IP toolkit to is because of its close relation to the lysosome, one of the organelles for which the lab had already built such a tool. This close relation proved relevant in Adelmann’s research on MFSD12, when he discovered that the protein is also required for the transport of cysteine into lysosomes. People with the rare genetic disorder cystinosis are affected by the buildup of cystine, another form of cysteine, in lysosomes. Adelmann found that by inhibiting MFSD12, and preventing cysteine from entering lysosomes, he could reverse the buildup of cystine in cells with the genetic mutation linked to cystinosis, suggesting a potential therapeutic use for MFSD12 inhibitors.
Adelmann is now turning his attention to cracking the identity of more of the proteins active in melanosomes and uncovering more of the biology underlying variation in skin color.
***
Written by Greta Friar
***
Adelmann, Charles H. et al. “MFSD12 mediates the import of cysteine into melanosomes and lysosomes.” Nature, Nov. 18, 2020. DOI: 10.1038/s41586-020-2937-x
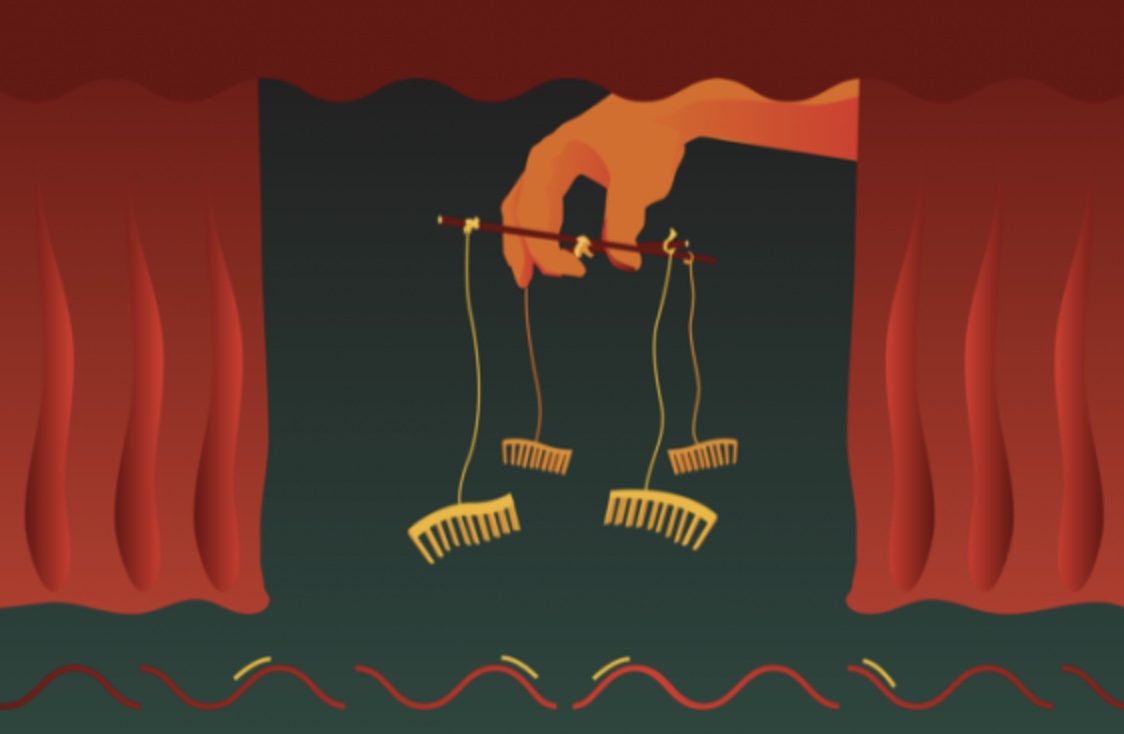
Whitehead Institute
November 12, 2020
MicroRNAs are short RNA sequences that maintain a tight control on which genes are expressed and when. They do this by regulating which messenger RNA (mRNA) transcripts — the single-stranded templates for proteins — are actually read by the cell. But what controls these cellular controllers?
In a new study published Nov. 12 in Science, researchers in David Bartel’s lab at Whitehead Institute show that mRNAs and other RNAs often turn the tables on their microRNA regulators — and show that the path to microRNA degradation is not what scientists expected it to be.
“A lot of people know that microRNAs repress mRNAs — that’s textbook,” said Charlie Shi, a graduate student in Bartel’s lab and first author on the paper. “But in certain cases, this logic is reversed. And I think that’s really interesting and weird, this idea that often the tables are turned.”
When transcripts attack
MicroRNAs typically control gene expression by binding to mRNA transcripts, and then working together with a protein called Argonaute to “silence” those transcripts by causing them to be more rapidly degraded. Because microRNAs are held cozily inside of the Argonaute protein, they are shielded from destructive enzymes in the cell, and are thus fairly long-lived by cellular standards. They can persist for up to a week, causing the destruction of many mRNA molecules over that time.
Sometimes, however, a microRNA binds to a special target site on an mRNA transcript that leads to premature destruction of the microRNA. This phenomenon — called target-directed microRNA degradation, or TDMD — happens naturally in cells, and is a way to control how much of certain microRNAs are allowed to persist at any given time.
Bartel’s lab began studying this form of degradation after researchers in the lab discovered that an RNA called CYRANO, which doesn’t code for any proteins, leads to the degradation of a specific microRNA called miR-7. This interaction was interesting to the researchers because the mechanism did not seem to line up with the current theories about TDMD.
Previous models of TDMD suggested that special target sites, like the one in CYRANO, cause one end of the microRNA to stick out of Argonaute and become vulnerable to the addition and subtraction of nucleotides by cytoplasmic enzymes. This process, called tailing and trimming, was thought to be a key step in the path to degradation of the microRNA.
“But when you knock out the enzyme that causes tailing of miR-7, it has no impact on the degradation,” Shi said. “So that’s curious, right? So how can we really perturb this supposedly responsible system and have no impact?”
A new model
In order to further probe the mechanism of TDMD, the researchers focused in on this interaction between the CYRANO noncoding RNA and miR-7. Shi designed a CRISPR screen to identify genes essential for the microRNA’s degradation when it encountered a CYRANO transcript.
The screen yielded one gene that was essential to the microRNA’s degradation, called ZSWIM8. When they looked up the gene’s function, the researchers found that it codes for a component of a ubiquitin ligase. Ubiquitin — so named because it is found in virtually all types of cells — serves as a flag to mark proteins for degradation in a cellular garbage disposal called the proteasome.
The finding of the ZSWIM8 ubiquitin ligase implied that CYRANO-mediated microRNA degradation involves destruction of the Argonaute protein. In this new molecular model for TDMD, the regulating RNA, CYRANO, binds to the microRNA, mir-7, encased in its protective Argonaute protein, and then recruits the ZSWIM8 ubiquitin ligase. This ligase then sticks a few ubiquitin molecules onto the microRNA’s Argonaute, leading Argonaute to be degraded, and thereby exposing its microRNA cargo to be destroyed by enzymes in the cell. Importantly, this process does not require any trimming and tailing of the microRNA.
“The discovery of this unanticipated pathway for TDMD illustrates the power of CRISPR screens, which can simultaneously query essentially every protein in the cell, including those that you never dreamed would be involved,” said Bartel, who is also an investigator of the Howard Hughes Medical Institute and a professor of biology at Massachusetts Institute of Technology.
A multitude of microRNAs
When the researchers looked at other known examples of TDMD, they found the ZSWIM8 was essential in all of them. Having identified this key part of the degradation pathway allowed them to seek out more microRNAs that are subject to this regulation.
“When we started this project, there were only around four examples in nature of endogenous RNAs that are encoded by the cell that can perform TDMD,” Shi said. “We had a feeling that there would be many more, and so by finding a factor that was required for TDMD in a general way — ZSWIM8 –we were then able to ask and answer the question, ‘how widespread this phenomenon?’”
As it turns out, TDMD is fairly common in multicellular organisms. The researchers looked for evidence of the microRNA degradation mechanism in different cell types — two from mice, and one from fruit flies — and found that in any given cell, up to 20 different microRNAs were regulated by TDMD out of a couple hundred total microRNAs in the cell.
The researchers also observed this mechanism in human cells and nematodes, suggesting that TDMD as a method for regulating microRNAs dates back to the common ancestor of these disparate species. That definitely creates a lot of questions for us,” Shi said. “Each one of these microRNAs is a story.”

How an MIT Biology alum from Puerto Rico came to love living in Boston
Saima Sidik
October 27, 2020
Even as a kid, Amaris Torres-Delgado PhD ’16 was analytical. “I wanted to be fact-based,” she says. “Once I had the facts, I’d speak with conviction.” As a result, her family wasn’t surprised that she decided to earn a PhD from MIT Biology, then apply for jobs in the pharmaceutical industry. Now, she works as a process development scientist at Amgen, where she uses her analytical skills to optimize drug production.
Torres-Delgado grew up in Puerto Rico, and the people, mindsets — and even the food — that she encountered in Cambridge, Massachusetts were unfamiliar at first. But after a decade of living in the Boston area, Torres-Delgado has come to love her new home, and she embraces the diversity of people and scientific problems she encounters.

In high school, Torres-Delgado considered becoming either a medical doctor or a lawyer. But because Torres-Delgado loves problem solving, her mother suggested that she consider becoming a scientist instead. This advice led her to earn a bachelor’s degree in industrial biotechnology from the University of Puerto Rico at Mayaguez. The drug company Amgen helped create this degree program in order to train future employees for its Puerto Rican branch. Torres-Delgado found the program to be an exciting opportunity to learn a combination of biology, chemistry, and chemical engineering, as well as a doorway into a meaningful career in the pharmaceutical industry.
During college, Torres-Delgado spent a summer working in Tania Baker’s lab as part of the MIT Summer Research Program in Biology (MSRP-Bio). “The mentorship I received was wonderful,” she says, and so when she was accepted to the MIT Biology Graduate Program, she didn’t hesitate to return, and she opted to stay in the Baker lab.
Being more than a thousand miles from home left Torres-Delgado feeling lonely, but fortunately, another Puerto Rican graduate student introduced her to a new hobby: salsa dancing. “We’d go to socials at the different salsa schools around Boston,” Torres-Delgado says. With this new community, she started to feel less homesick.
In the lab, Torres-Delgado became captivated by a protein degradation machine that others in the Baker lab were studying. Cells use these wood-chipper-like machines to regulate protein levels, and a component of this machine called ClpS carries proteins to the site where they’re destroyed. Strikingly, ClpS speeds up the degradation of some kinds of proteins and slows down the degradation of others, but no one had been able to figure out why. Although other Baker lab members told Torres-Delgado that the ClpS mystery would be tricky to solve, she was determined to crack this cold case.
By the end of her PhD, she’d discovered that, in addition to delivering proteins to the degradation machine, ClpS sits on the same machine and makes it work less efficiently. Carrying certain proteins to the machine speeds up their degradation, but sitting on the machine slows down degradation of incoming proteins.
Although she enjoyed learning biochemistry in the Baker lab, Torres-Delgado says, “I’ve always been excited about pharmaceutical work that goes on close to the patient.” Her original plan was to return to Puerto Rico after earning her doctorate in order to work as an industry scientist there, but when she finished her PhD, she felt like she wasn’t done exploring Boston.

She took a job at Vertex Pharmaceuticals with a group that oversaw manufacturing of the company’s first drug based on a biological molecule. While many drugs are produced through chemical reactions, this drug was produced in living cells, and Torres-Delgado was part of the team that supervised this new area of drug production. The biochemistry she’d learned during her PhD gave her the scientific background to provide valuable insight, but Torres-Delgado had a lot to learn about the process of efficiently producing a high-quality drug, and her industry colleagues helped her pick up the new skills she needed.
“I learned these skills on the job, from my peers, and this way of learning is something that’s available and encouraged,” she says. “You don’t have to be super focused on your long-term career goals during your training.” She’s since moved to Amgen’s Cambridge branch, where she works in process development as part of their oncology division.
Ten years after leaving her childhood home in Puerto Rico, Torres-Delgado still doesn’t feel like she’s finished living in Boston. She moved north at an impressionable point in her life, at a time when minority rights were gaining traction, and the people and philosophies she found in Boston have impacted her world view substantially.
“As a young adult, I wanted to experience a way of living that differed from how I grew up,” she says. “I didn’t realize how much more there is to the world until I moved to Boston. Here, I’ve had the opportunity to learn about other religions, other cultures, people from the whole gender spectrum — even understanding that there is a gender spectrum was a new experience.”
Torres-Delgado also finds diversity in her job, which includes a variety of tasks like figuring out how to optimize a manufacturing process, making sure Amgen meets regulatory standards, and mentoring other scientists. Underlying all these skills is the same analytical mindset that she started developing back in Puerto Rico and built on at MIT — it’s all about leveraging the facts.
Posted 10.22.20
Top photo: Amaris Torres-Delgado/Ammar Arsiwala

Greta Friar | Whitehead Institute
September 17, 2020
Tiny mitochondria in our cells turn oxygen and nutrients into usable energy in a process called respiration. This process is essential for powering our cells, and yet in spite of its importance many of the finer details of how it happens remain unknown. One long-standing mystery is how a molecule called nicotinamide adenine dinucleotide (NAD), which plays a big part in respiration and metabolism, gets into the mitochondria in humans and other animals. Mitochondria use NAD in order to produce adenosine triphosphate (ATP), the energy supply molecules used throughout the cell. Researchers knew the identities of the molecules that transport NAD from the wider cell into the mitochondria of yeast and plants, but had not found the animal equivalent—in fact, there was some debate over whether one even existed or whether animal cells used other methods altogether.
Now, research from postdoctoral researcher Nora Kory in Whitehead Institute Member David Sabatini’s lab may end the debate. In a paper published in Science Advances on September 9, the researchers show that the missing human NAD transporter is likely the protein MCART1. This discovery not only answers a longstanding question about a vital cellular process, but may contribute to research on aging—during which cells’ NAD levels drop—as well as research on diseases that involve certain mitochondrial dysfunctions, for which cells with broken NAD transporters could be an experimental model.
“I find it striking that mitochondria play such an important role in metabolism in the cell, which in turn plays a huge role in health and disease, but we still don’t understand how all of the molecules involved get in and out of mitochondria. It was exciting to fill in a piece of that puzzle.” Kory says.
AN UNEXPECTED DISCOVERY
Kory did not set out to find the long sought-after transport molecule. Rather, she was trying to better understand mitochondrial respiration by mapping the genes involved. She was comparing gene essentiality profiles, which show how important a gene is to different processes in a cell—the more co-essential two genes are, the more likely they are to be involved in the same cellular process—and one gene stood out: MCART1, also known as SLC25A51. It was highly correlated to other genes involved in mitochondrial respiration, and belonged to a family of genes known to code for transporters, yet its function was unknown. The protein coded for by MCART1 clearly played an important role, so Kory decided to figure out what that was; as her research progressed, she realized she had found the missing NAD transporter.
Kory and colleagues applied a common approach to determine MCART1’s function: inactivate the gene in cells, and see what breaks down in its absence. This approach is like troubleshooting a machine; if you cut a wire in your car and the headlights stop working, but everything else is fine, then that wire was probably linked to the headlights. When the researchers removed MCART1, the cells exhibited much lower oxygen consumption, reduced respiration and ATP production, and reliance on other, far less efficient means of ATP production—exactly what you’d expect to see if the inactivated gene was needed for respiration. Moreover, the biggest change that the researchers observed in cells without MCART1 was reduced levels of NAD in the mitochondria, while NAD levels in the wider cell remained the same, which they quantified using experiments previously developed in the lab. The researchers confirmed that MCART1 is essential for NAD transport into isolated mitochondria and overabundance of MCART1 caused an increased uptake.
“It’s very satisfying when our lab returns to the techniques that we have developed in order to make new findings such as identifying this important protein,” says Sabatini, who is also a professor of biology at Massachusetts Institute of Technology and an investigator with the Howard Hughes Medical Institute.
The evidence supports that the protein MCART1 is itself the transport channel. However, it is possible that the protein may play some other essential contributing role to transportation, or that it combines with other molecules to do its job. To strengthen the case for MCART1 as the transporter, the researchers showed that MCART1 and the known yeast NAD transport could be switched out for each other in both human and yeast cells, suggesting an equivalent function. Still, further experiments are needed to determine the precise mechanism of transport.
A serendipitous case of synchronous discovery reinforces Kory’s findings. A paper by other researchers published on the same day in the journal Nature also put forth that MCART1 is the missing NAD transporter, based on a completely different set of evidence. Combined, the papers provide an even more compelling case.
“It was nice to see how our different approaches complemented each other, and led to the same conclusion,” Kory says.
Understanding how NAD gets into the mitochondria opens up new questions about the details of mitochondrial respiration. Kory will shortly be leaving Sabatini’s lab to open her own lab at the Harvard T.H. Chan School of Public Health, where she intended to continue investigating the role of the mitochondria’s NAD supply in metabolism and signaling.
***
Written by Greta Friar
***
David Sabatini’s primary affiliation is with Whitehead Institute for Biomedical Research, where his laboratory is located and all his research is conducted. He is also a Howard Hughes Medical Institute investigator and a professor of biology at Massachusetts Institute of Technology.
***
Citations:
Kory, N., et al. (2020). MCART1/SLC25A51 is required for mitochondrial NAD transport. Science Advances. doi:10.1126/sciadv.abe5310
Luongo, T. S., et al. (2020). SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature. doi:10.1038/s41586-020-2741-7
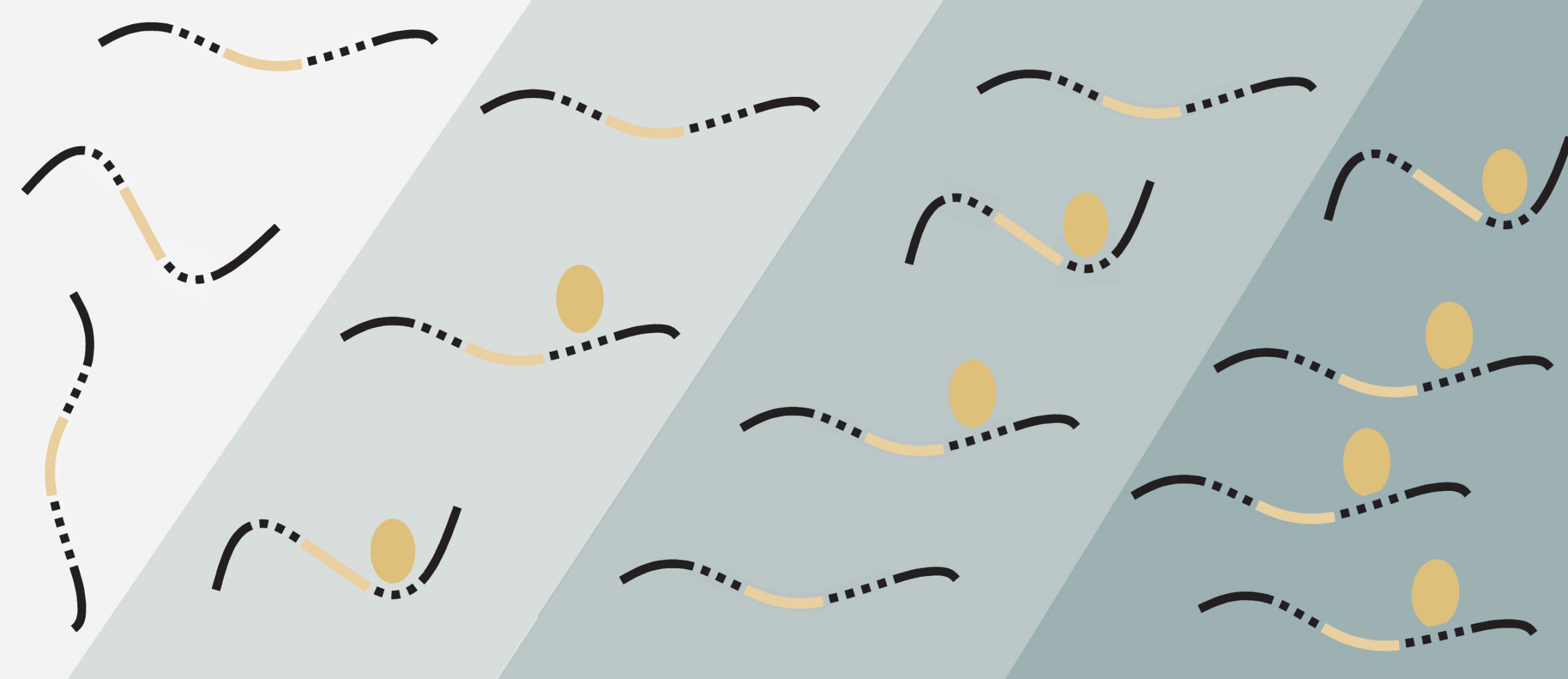
A well-known protein family binds to many more RNA sequences than previously thought to help neurons grow.
Raleigh McElvery
August 17, 2020
In every cell, RNA-binding proteins (RBPs) help tune gene expression and control biological processes by binding to RNA sequences. Researchers often assume that individual RBPs latch tightly to just one RNA sequence. For instance, an essential family of RBPs, the Rbfox family, was thought to bind one particular RNA sequence alone. However, it’s becoming increasingly clear that this idea greatly oversimplifies Rbfox’s vital role in development.
Members of the Rbfox family are among the best-studied RBPs and have been implicated in mammalian brain, heart, and muscle development since their discovery 25 years ago. They influence how RNA transcripts are “spliced” together to form a final RNA product, and have been associated with disorders like autism and epilepsy. But this family of RBPs is compelling for another reason as well: until recently, it was considered a classic example of predictable binding.
More often than not, it seemed, Rbfox proteins bound to a very specific sequence, or motif, of nucleotide bases, “GCAUG.” Occasionally, binding analyses hinted that Rbfox proteins might attach to other RNA sequences as well, but these findings were usually discarded. Now, a team of biologists from MIT has found that Rbfox proteins actually bind less tightly — but no less frequently — to a handful of other RNA nucleotide sequences besides GCAUG. These so-called “secondary motifs” could be key to normal brain development, and help neurons grow and assume specific roles.
“Previously, possible binding of Rbfox proteins to atypical sites had been largely ignored,” says Christopher Burge, professor of biology and the study’s senior author. “But we’ve helped demonstrate that these secondary motifs form their own separate class of binding sites with important physiological functions.”
Graduate student Bridget Begg is the first author of the study, published on August 17 in Nature Structural & Molecular Biology.
“Two-wave” regulation
After the discovery that GCAUG was the primary RNA binding site for mammalian Rbfox proteins, researchers characterized its binding in living cells using a technique called CLIP (crosslinking-immunoprecipitation). However, CLIP has several limitations. For example, it can indicate where a protein is bound, but not how much protein is bound there. It’s also hampered by some technical biases, including substantial false-negative and false-positive results.
To address these shortcomings, the Burge lab developed two complementary techniques to better quantify protein binding, this time in a test tube: RBNS (RNA Bind-n-Seq), and later, nsRBNS (RNA Bind-n-Seq with natural sequences), both of which incubate an RBP of interest with a synthetic RNA library. First author Begg performed nsRBNS with naturally-occurring mammalian RNA sequences, and identified a variety of intermediate-affinity secondary motifs that were bound in the absence of GCAUG. She then compared her own data with publicly-available CLIP results to examine the “aberrant” binding that had often been discarded, demonstrating that signals for these motifs existed across many CLIP datasets.
To probe the biological role of these motifs, Begg performed reporter assays to show that the motifs could regulate Rbfox’s RNA splicing behavior. Subsequently, computational analyses by Begg and co-author Marvin Jens using mouse neuronal data established a handful of secondary motifs that appeared to be involved in neuronal differentiation and cellular diversification.
Based on analyses of these key secondary motifs, Begg and colleagues devised a “two-wave” model. Early in development, they believe, Rbfox proteins bind predominantly to high-affinity RNA sequences like GCAUG, in order to tune gene expression. Later on, as the Rbfox concentration increases, those primary motifs become fully occupied and Rbfox additionally binds to the secondary motifs. This results in a second wave of Rbfox-regulated RNA splicing with a different set of genes.
Begg theorizes that the first wave of Rbfox proteins binds GCAUG sequences early in development, and she showed that they regulate genes involved in nerve growth, like cytoskeleton and membrane organization. The second wave appears to help neurons establish electrical and chemical signaling. In other cases, secondary motifs might help neurons specialize into different subtypes with different jobs.
John Conboy, a molecular biologist at Lawrence Berkeley National Lab and an expert in Rbfox binding, says the Burge lab’s two-wave model clearly shows how a single RBP can bind different RNA sequences — regulating splicing of distinct gene sets and influencing key processes during brain development. “This quantitative analysis of RNA-protein interactions, in a field that is often semi-quantitative at best, contributes fascinating new insights into the role of RNA splicing in cell type specification,” he says.
A binding spectrum
The researchers suspect that this two-wave model is not unique to Rbfox. “This is probably happening with many different RBPs that regulate development and other dynamic processes,” Burge says. “In the future, considering secondary motifs will help us to better understand developmental disorders and diseases, which can occur when RBPs are over- or under-expressed.”
Begg adds that secondary motifs should be incorporated into computer models that predict gene expression, in order to probe cellular behavior. “I think it’s very exciting that these more finely-tuned developmental processes, like neuronal differentiation, could be regulated by secondary motifs,” she says.
Both Begg and Burge agree it’s time to consider the entire spectrum of Rbfox binding, which are highly influenced by factors like protein concentration, binding strength, and timing. According to Begg, “Rbfox regulation is actually more complex than we sometimes give it credit for.”
Citation:
“Concentration-dependent splicing is enabled by Rbfox motifs of intermediate affinity”
Nature Structural & Molecular Biology, online August 17, 2020, DOI: 10.1038/s41594-020-0475-8
Bridget E. Begg, Marvin Jens, Peter Y. Wang, Christine M. Minor, and Christopher B. Burge