Bailey Bowcutt investigated COVID-19 cases in rural Wyoming before coming to MIT for the summer and applying her knowledge to a new cellular invader.
Raleigh McElvery
July 23, 2021
The first time Bailey Bowcutt saw a lab it was nothing like she expected. Rather than a stark, sterile setting with sullen figures floating around like ghosts in white lab coats, the atmosphere was cordial and the dress casual. Some scientists even sported vibrant shirts with Marvel characters. A high school senior on a class field trip, Bowcutt couldn’t have predicted that the next time she’d set foot in the Wyoming Public Health Laboratory she’d no longer be a visitor, but a researcher performing diagnostic testing during a global pandemic. Now, as COVID-19 restrictions begin to lift, she’s taking the research tools she’s learned to Cambridge, Massachusetts to complete the Bernard S. and Sophie G. Gould MIT Summer Research Program in Biology (BSG-MSRP-Bio) and investigate how other types of pathogens spread.
Growing up in rural Wyoming, Bowcutt had little exposure to science because there were few research institutes close by. But watching family members suffer from gastrointestinal illness and other infections spurred her to pursue a degree in microbiology at Michigan State University (MSU). Shortly after she arrived on campus in the fall of 2019, she joined Shannon Manning’s lab studying antibiotic resistance in cattle.
Cows are prone to contracting a bacterial infection of the udder called mastitis. (In humans, a similar inflammation can occur in breast tissue.) Manning’s lab is looking at how antibiotic treatments affect the bovine gut microbiome and emergence of antibiotic resistance genes. Bowcutt’s role was to help identify these super bugs inside the cows’ gastrointestinal tracts.
“I got to go to the farm to take samples, which involved a glove that goes all the way up to the shoulder and some invasive maneuvers inside cows,” she explains. “Luckily, I was just the bag holder!”
Intimate sample collection aside, Bowcutt was excited about the work because it combined agriculture and human health research to solve issues plaguing rural communities. But her time on the farm was cut short when COVID-19 cases climbed in early 2020. She headed back to her home in Wyoming to begin remote MSU classes and, reminiscing about her field trip to the Wyoming Public Health Laboratory, reached out to the director to see if there were any internship opportunities.
“I’d barely learned how to do science at that point, but they needed people who could handle a pipette, so they took me,” she says. “I ended up being one of the first people there helping with COVID research, and I stayed for about a year-and-a-half while I took online classes.”
The lab would receive nasopharyngeal swabs from COVID-19 patients, and Bowcutt’s first task was to help extract RNA from the samples. Later, she transitioned to another project, which required performing PCR on untreated wastewater samples to glean a population-level understanding of where COVID-19 outbreaks were occurring.
She began toying with the idea of pursuing a PhD, but wasn’t sure what it would entail. So, in early 2021, she started Googling summer science programs and stumbled on BSG-MSRP-Bio. She was accepted, and paired with one of the very labs that had caught her eye online: assistant professor Becky Lamason’s group.
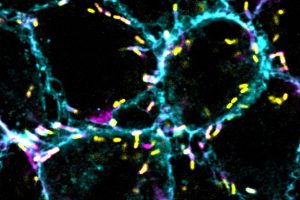
“If you’ve ever seen microscopy pictures from the Lamason lab, they’re just so beautiful,” Bowcutt explains. Beautiful, yes — but she would soon learn these snapshots capture a chilling cellular invasion and molecular heist.
The Lamason lab watches malicious bacteria as they hijack molecules in human host cells to build long tails, rocket around, and punch through the cell membrane to spread. Bowcutt’s mentor, graduate student Yamilex Acevedo-Sánchez, focuses on the food-borne bacterium Listeria monocytogenes, which targets the gastrointestinal tract. Acevedo-Sánchez’s research aims to understand the host cell pathways that Listeria commandeers to move from one cell to the next in a process called cell-to-cell spread.
Together, Acevedo-Sánchez and Bowcutt are investigating several proteins in the human host cell involved in cellular transport and membrane remodeling (Caveolin-1, Pacsin2, and Fes), which could regulate Listeria’s spread. Over the summer, the duo has been adjusting the levels of these proteins and observing what happens to Listeria’s ability to move from cell-to-cell.
Bowcutt spends most of her days doing Western blots; growing Listeria and mammalian cells; and combining immunofluorescence assays with fixed and live cell microscopy to take her own striking microscopy images and movies of the parasites.
“I expected the work environment at MIT to be very intense, but everyone has been really friendly and willing to answer questions,” she says. “Some of my favorite experiences have just been in the lab while everyone is bustling around. It’s a welcome change after so much COVID-19 isolation.”
Now that the COVID-era occupancy restrictions have lifted, Bowcutt’s lab bench neighbor is Lamason herself. “She’s next to me doing experiments all the time,” Bowcutt explains, “which is cool because she’s really engaging with the research in the same way we are.”
Bowcutt says her summer experience has given her some much-needed practice designing research questions and devising the experiments to answer them. She’s also acquired a new skill she didn’t anticipate: interpreting ambiguous results and developing follow-up experiments to clarify them.
These days, the prospect of a PhD seems much less intimidating. In fact, the Lamason lab has done more than simply pique Bowcutt’s interested in fundamental biology research. She’s now considering ways to combine her microbiology skills with her interest in rural health care.
“I didn’t expect to see this much growth in myself,” she says, “and I know it’s making me a better scientist. I’m excited to return to MSU in the fall because I feel like I can do so much more now — and I would totally do it again.”