Cells rely on complex molecular machines composed of protein assemblies to perform essential functions such as energy production, gene expression, and protein synthesis. To better understand how these machines work, scientists capture snapshots of them by isolating proteins from cells and using various methods to determine their structures. However, isolating proteins from cells also removes them from the context of their native environment, including protein interaction partners and cellular location.
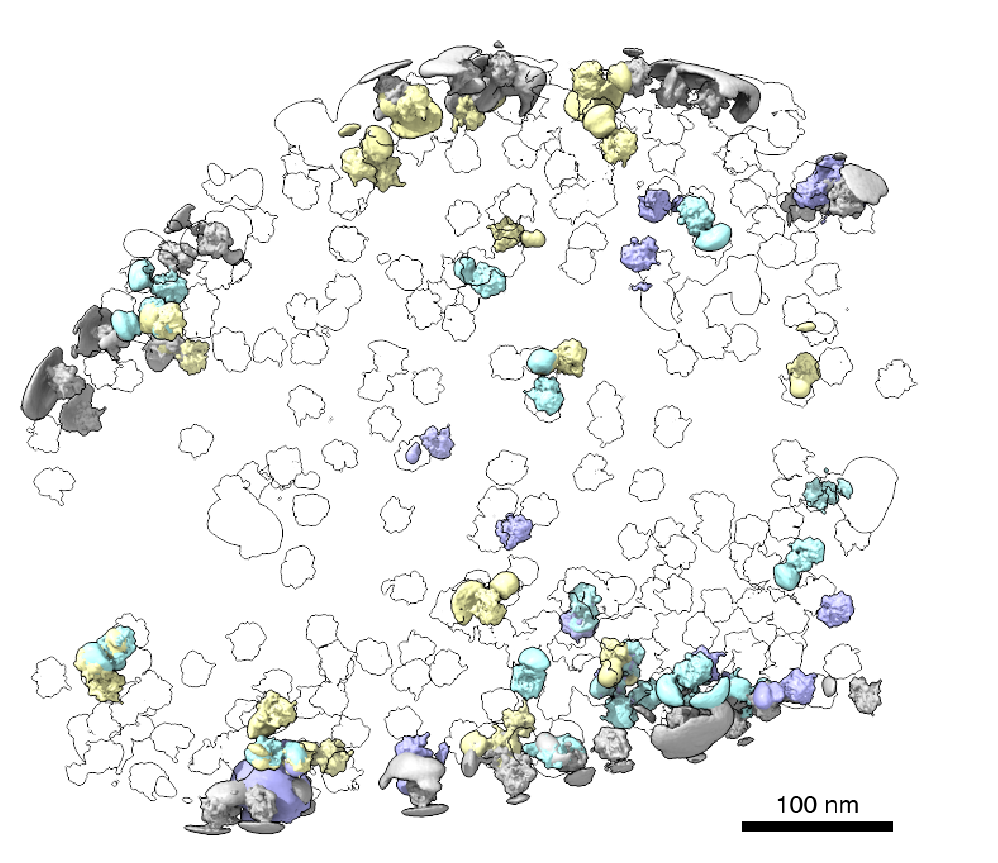
Recently, cryogenic electron tomography (cryo-ET) has emerged as a way to observe proteins in their native environment by imaging frozen cells at different angles to obtain three-dimensional structural information. This approach is exciting because it allows researchers to directly observe how and where proteins associate with each other, revealing the cellular neighborhood of those interactions within the cell.
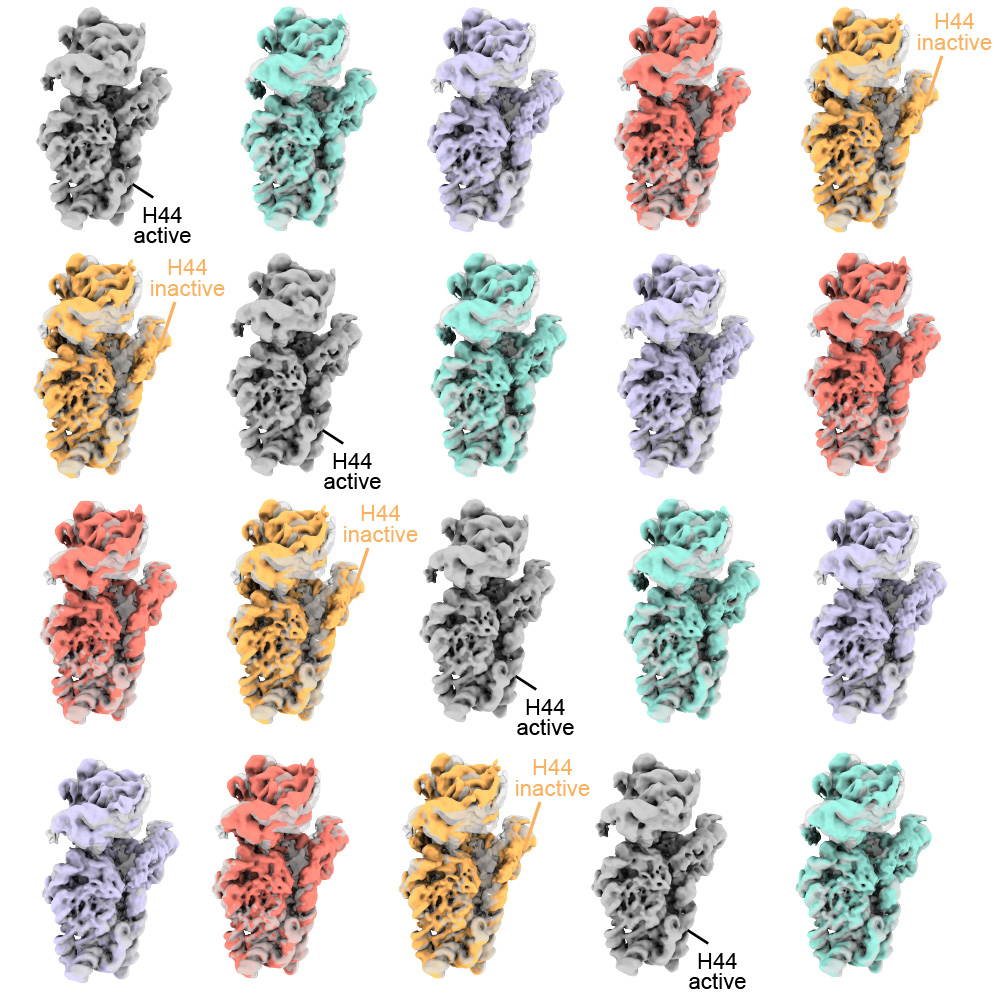
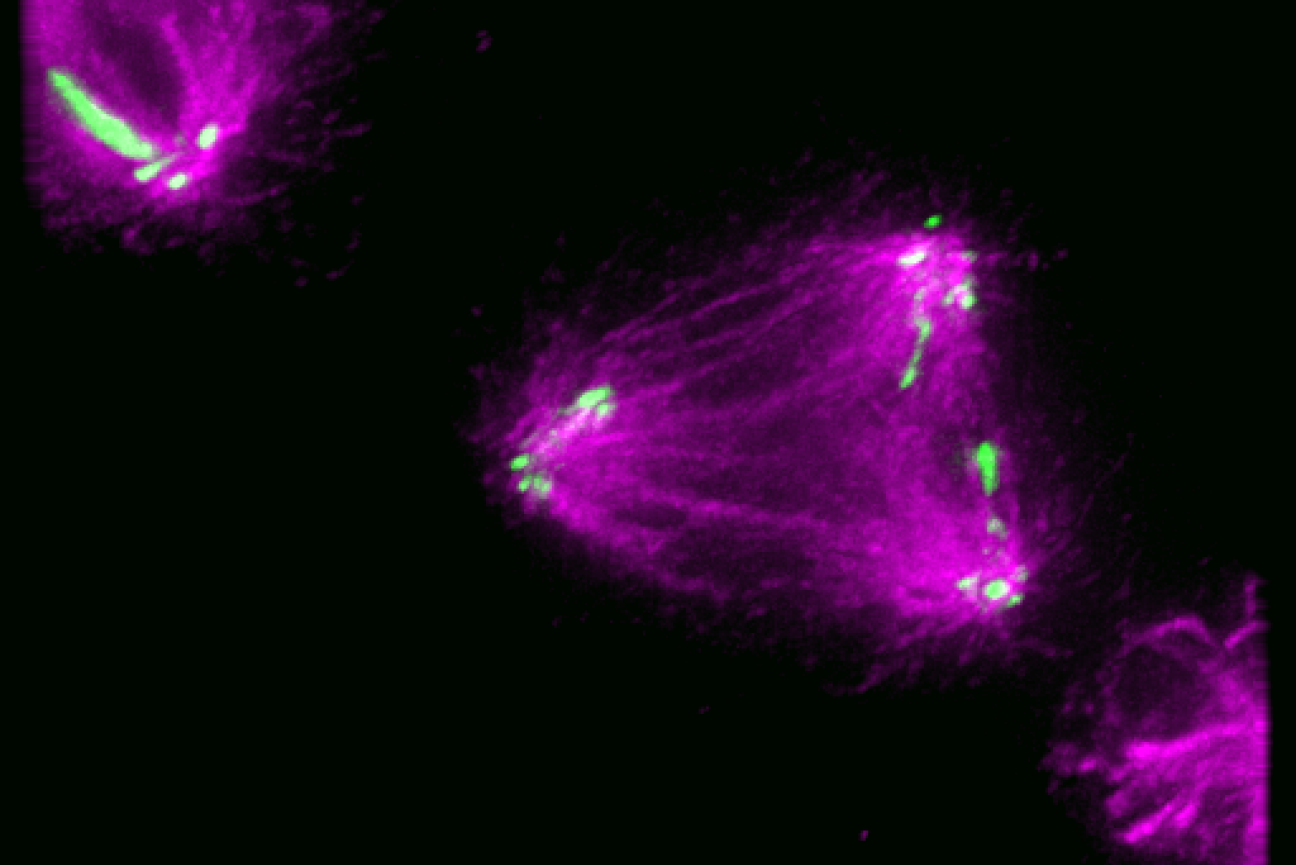
With the technology available to image proteins in their native environment, graduate student Barrett Powell wondered if he could take it one step further: what if molecular machines could be observed in action? In a paper published today in Nature Methods, Powell describes the method he developed, called tomoDRGN, for modeling structural differences of proteins in cryo-ET data that arise from protein motions or proteins binding to different interaction partners. These variations are known as structural heterogeneity.
Although Powell had joined the Davis Lab as an experimental scientist, he recognized the potential impact of computational approaches in understanding structural heterogeneity within a cell. Previously, the Davis Lab developed a related methodology named cryoDRGN to understand structural heterogeneity in purified samples. As Powell and Associate Professor of Biology Joey Davis saw cryo-ET rising in prominence in the field, Powell took on the challenge of reimagining this framework to work in cells.
When solving structures with purified samples, each particle is imaged only once. By contrast, cryo-ET data is collected by imaging each particle more than 40 times from different angles. That meant tomoDRGN needed to be able to merge the information from more than 40 images, which was where the project hit a roadblock: the amount of data led to an information overload.
To address the information overload, Powell successfully rebuilt the cryoDRGN model to prioritize only the highest-quality data. When imaging the same particle multiple times, radiation damage occurs. The images acquired earlier, therefore, tend to be of higher quality because the particles are less damaged.
“By excluding some of the lower quality data, the results were actually better than using all of the data–and the computational performance was substantially faster,” Powell says.
Just as Powell was beginning work on testing his model, he had a stroke of luck: the authors of a groundbreaking new study that visualized, for the first time, ribosomes inside cells at near-atomic resolution, shared their raw data on the Electric Microscopy Public Image Archive (EMPIAR). This dataset was an exemplary test case for Powell, through which he demonstrated that tomoDRGN could uncover structural heterogeneity within cryo-ET data.
According to Powell, one exciting result is what tomoDRGN found surrounding a subset of ribosomes in the EMPIAR dataset. Some of the ribosomal particles were associated with a bacterial cell membrane and engaged in a process called cotranslational translocation. This occurs when a protein is being simultaneously synthesized and transported across a membrane. Researchers can use this result to make new hypotheses about how the ribosome functions with other protein machinery integral to transporting proteins outside of the cell, now guided by a structure of the complex in its native environment.
After seeing that tomoDRGN could resolve structural heterogeneity from a structurally diverse dataset, Powell was curious: how small of a population could tomoDRGN identify? For that test, he chose a protein named apoferritin which is a commonly used benchmark for cryo-ET and is often treated as structurally homogeneous. Ferritin is a protein used for iron storage and is referred to as apoferritin when it lacks iron.
Surprisingly, in addition to the expected particles, tomoDRGN revealed a minor population of ferritin particles–with iron bound–making up just 2% of the dataset that was not previously reported. This result further demonstrated tomoDRGN’s ability to identify structural states that occur so infrequently that they would be averaged out with traditional analysis tools.
Powell and other members of the Davis Lab are excited to see how tomoDRGN can be applied to further ribosomal studies and to other systems. Davis works on understanding how cells assemble, regulate, and degrade molecular machines, so the next steps include exploring ribosome biogenesis within cells in greater detail using this new tool.
“What are the possible states that we may be losing during purification?” Davis says. “Perhaps more excitingly, we can look at how they localize within the cell and what partners and protein complexes they may be interacting with.”
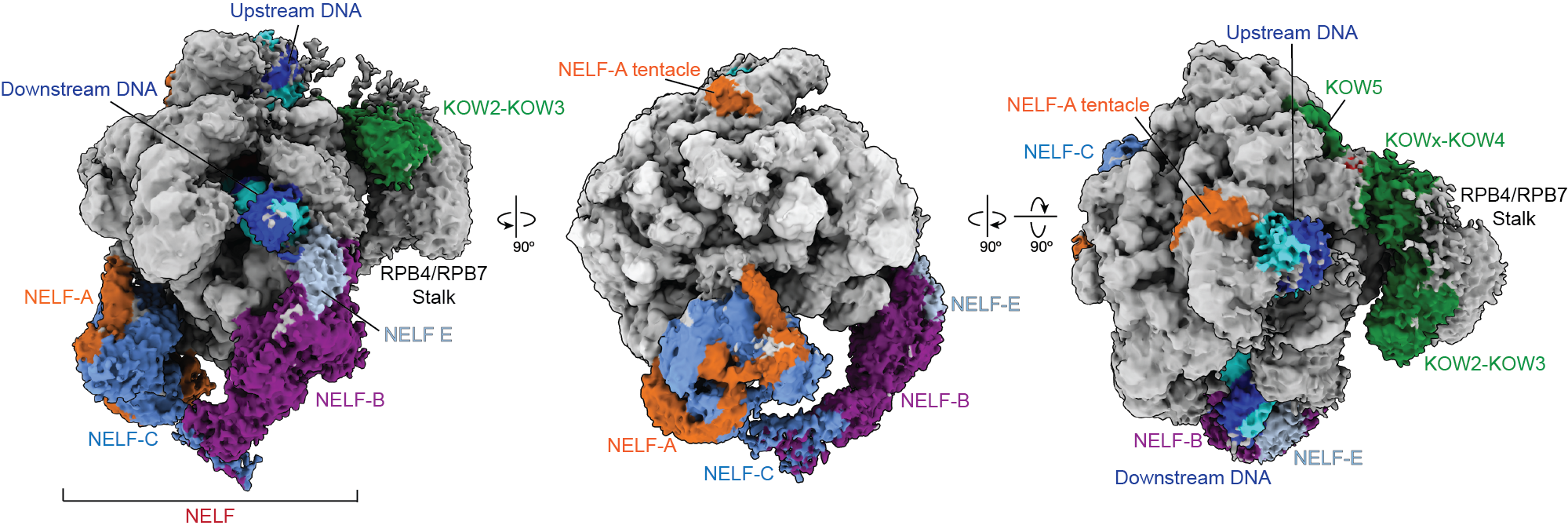
Transcription, the process of copying RNA from DNA, is a critical first step for cells to create proteins. The enzyme responsible for transcription is a motor protein called RNA polymerase.
When an RNA polymerase transcribes a gene, it will begin elongating the mRNA and will then, often, pause.
From there, the RNA polymerase will either return to elongating the mRNA or it will get stuck. For the latter occurrence, the mRNA and subsequent protein will never be made: the polymerase will go somewhere else, or restart transcription on the same gene and get stuck again.
Pausing is thought to be governed by a protein called Negative Elongation Factor, NELF, and DRB-sensitivity inducing factor, DSIF. Previous research suggested that NELF stably clamps down onto RNA polymerase to stall the elongation process and prevent the polymerase from moving. That model contradicted cell-based experiments, however, which indicated that NELF is somehow still attached to polymerase after transcription resumes.
New research from the Vos Lab in the Department of Biology at MIT published today in Molecular Cell reveals that NELF isn’t merely an on-off switch for transcription. Instead, NELF can change into a distinct conformation that allows the polymerase to resume transcription. The researchers dubbed this distinct conformation NELF’s “poised” state.
RNA polymerase pausing, sometimes for minutes at a time, is thought to be an important gene expression checkpoint; more than half of genes exhibit pausing, although many questions remain about the role of pausing in gene expression. Understanding both how and why the process is occurring, down to the atomic level, and what components are involved, is key to understanding how cells function, both individually and as part of an organism.
“It’s a very central question to biological research, and we still don’t fully understand it because it’s such a complex process,” says first author Bonnie G. Su, a graduate student in the Vos lab. “The bigger picture is: how does the cell decide what resources to allocate to certain biological processes? This finding might help us answer questions like that.”
To visualize the two distinct conformations of NELF and polymerase, the researchers used a combination of biochemical and structural approaches. The previous understanding of proximal pausing was based on Cryo-Electron Microscopy images of the static complex. Cryo-EM is a powerful microscopy technique that involves freezing samples and imaging them, and that approach had captured polymerase in its paused state.
Using the core Cryo-EM facility available at MIT.nano, Su instead added the necessary components for the polymerase to transcribe, and gathered structural data on an actively transcribing complex —allowing, for the first time, a stepwise visualization of how proximal pausing occurs.
“What we found is that NELF, which we always thought of as static, can actually move around,” Su says. “This has updated our understanding of what pausing is, and how early gene regulation happens.”
The structural results also provide an explanation for how polymerase may be cycling between the two states—and how one form of NELF may be forcing polymerase to pause, while the newly discovered form allows polymerase to resume transcription.
It’s still unclear what triggers NELF to transition from the paused state to the poised state, and many questions remain about how polymerase is regulated, according to senior author Seychelle Vos, the Robert A. Swanson (1969) Career Development Professor of Life Sciences and HHMI Freeman Hrabowski Scholar. RNA polymerase can be associated with and is known to be regulated by a large repertoire of proteins.
“We’re trying to see if we can actually lock the complex in the paused state by adding additional factors,” Vos says. “We’re also pursuing whether sequence context is affecting pausing behavior—how or if the sequence of DNA may be causing polymerase to pause.”
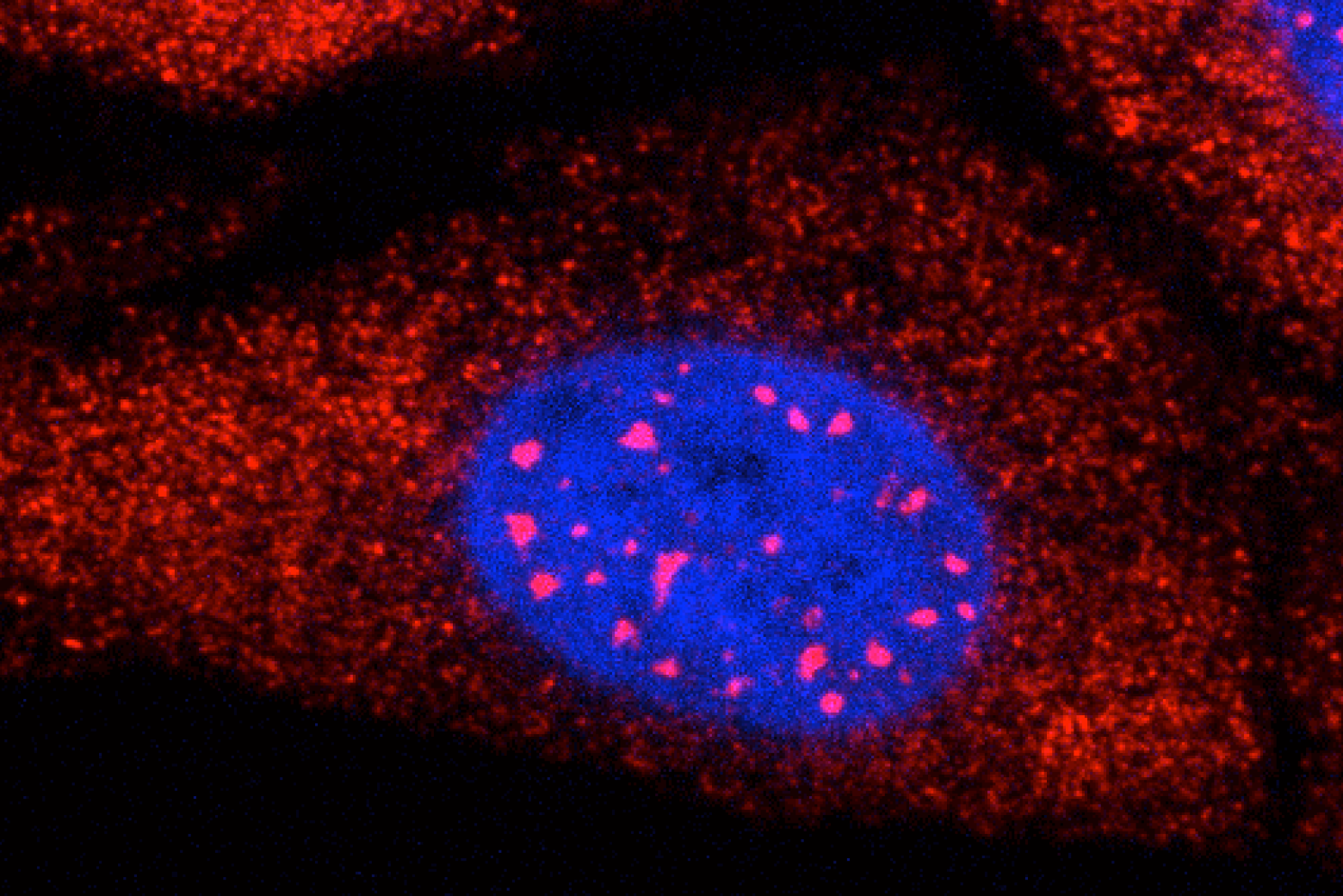
Retroviruses cannot replicate on their own — they must insert their genetic code into the DNA of a host and exploit the host cell’s resources to make more copies of themselves, furthering infection. Some retroviruses only infect cells as they divide, when the nuclear envelope that protects the host’s genetic material breaks down, making it easily accessible. HIV-1 is a type of retrovirus, called a lentivirus, that can infect non-dividing cells.
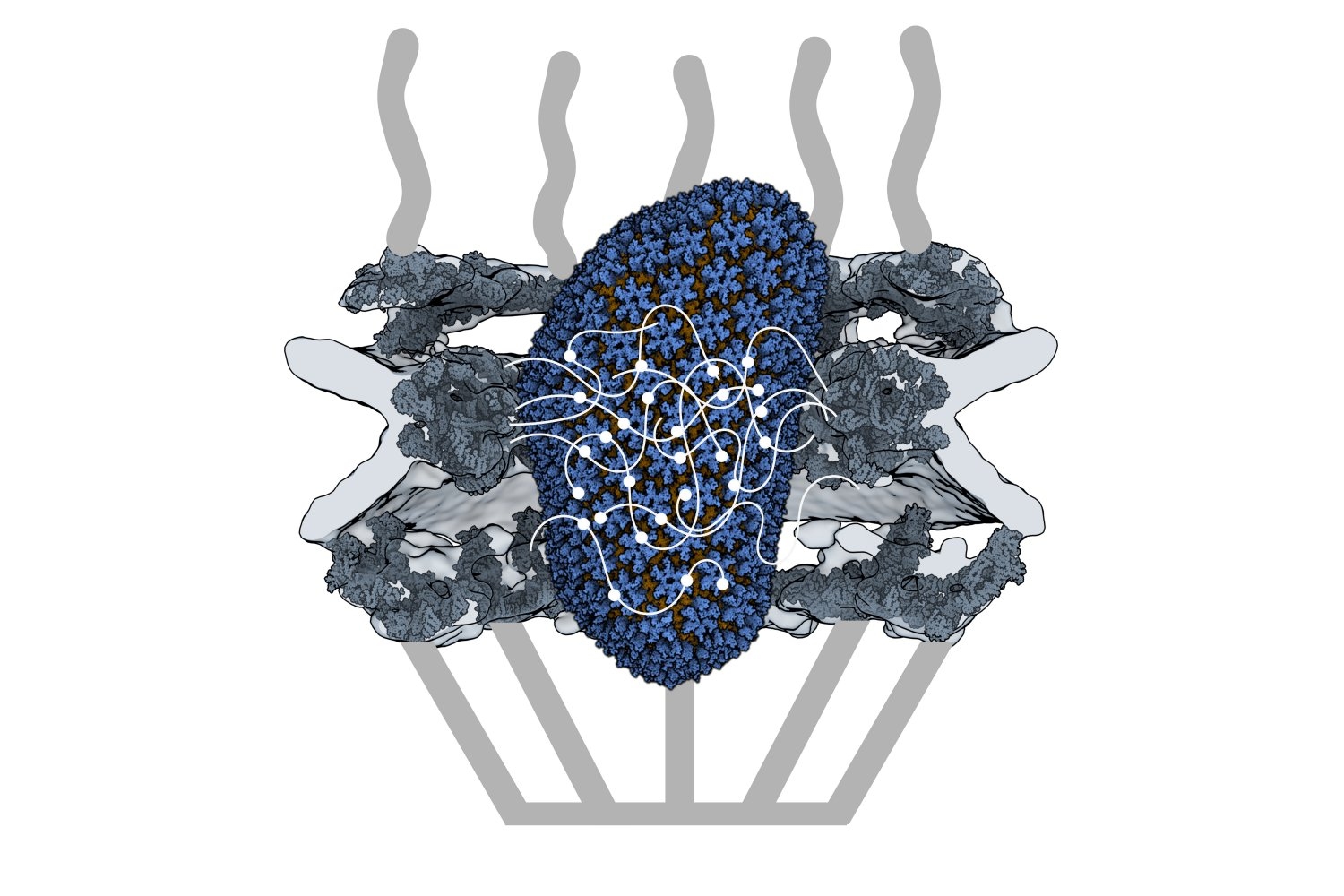
HIV-1 delivers its genome into the nucleus by packaging it into a large, cone-shaped structure called a capsid — but the exact mechanism has remained elusive for decades. Travel through the nuclear envelope occurs through, and is regulated by, nuclear pores, doughnut-shaped protein assemblies. Human cells have about 2,000 nuclear pores perforating the nuclear envelope. Some earlier evidence suggested that the capsid remains intact during its delivery into the nucleus — but this created a dimensional conundrum. The cone-shaped HIV-1 capsid is about 120 nanometers long and 60 nm wide — too large, researchers thought, to fit through the opening of the nuclear pore, measured at only 43 nm wide.
Members of the Schwartz Lab at MIT, in the Department of Biology, became interested in this question when a postdoc in the lab used cryo-electron tomography, slicing up sections of frozen cells to examine structures, to show that nuclear pores in the nuclear envelope are larger than 43nm. They deflate and shrink, it turns out, when removed from their native conditions. In native conditions, the nuclear pore complex is about 60nm wide — wide enough to accommodate the HIV-1 capsid.
Knowing that it could fit, a question remained: How can the capsid navigate the dense mesh of spaghetti-like proteins that act like a sieve in the nuclear pore channel? That spaghetti-like mesh allows small cargo to diffuse through, but prevents large cargo from entering unless it is escorted by proteins called nuclear transport receptors.
In an open-access paper published today in Nature, researchers present evidence that the HIV-1 capsid mimics the cell’s transport receptors to traverse the nuclear pore.
To support that conclusion, the researchers showed three things in vitro: that an HIV-1 capsid can deliver cargo through a nuclear pore analog; that the capsid can interact with the sieve of proteins in the nuclear pore channel; and that the capsid targets the nuclear pore in the absence of native transport proteins.
Nuclear transport receptors escort large cargo through the nuclear pore by “batting away” the spaghetti-like mesh of proteins inside the channel — like someone holding your hand and guiding you across a crowded dance floor. The HIV-1 capsid interacts with the spaghetti-like proteins, but its purpose is more like a Trojan horse — the capsid encapsulates the viral cargo, protecting it from detection in the cytoplasm and as it enters the nuclear pore complex.
“What’s really amazing about cells is that they are incredibly complex. What’s really difficult about studying cells is that they are incredibly complex,” jokes co-first author Erika Weiskopf, a graduate student in the Schwartz lab. “Biochemists are constantly trying to find ways to study their system in a simplified context, but still give it a flavor of cell biology.”
To do that, the Schwartz lab collaborated with Dirk Görlich, the director of cellular logistics at the Max Planck Institute for Multidisciplinary Sciences. Görlich is a co-senior author on the paper with MIT’s Boris Magasanik Professor of Biology Thomas Schwartz. Görlich’s lab has produced concentrated droplets of the spaghetti-like proteins found inside the nuclear pore, and those droplets allow and exclude cargo the same way a nuclear pore will. In experiments, fluorescently-labeled cargo did not enter the droplets, but fluorescently-labeled cargo packaged in an HIV-1 capsid was delivered. This indicated that the capsid could deliver cargo through a nuclear pore.
Using a biophysical binding assay, the researchers also showed that the HIV-1 capsid interacts with the proteins inside the channel. Different spaghetti-like proteins are found in different channel sections, such as at the cytoplasmic side’s entrance or only inside the channel; there are 10 such proteins in human cells. The capsid is a promiscuous binder — it can interact with all the spaghetti-like proteins found in the channel.
The capsid can target the nuclear pore complex even without the cell’s transport receptors, indicating that it is not commandeering native transport receptors to find and enter the nuclear pore. The team used a classic assay in the nucleocytoplasmic transport field to collect this evidence: When cells are treated with digitonin, their membranes become porous. Everything in the cytoplasm will leak out of the cells, but the nuclear envelope will remain intact. Despite the absence of native proteins, the capsid was attracted to the nuclear pore complex, a behavior indicative of a nuclear transport receptor.
Although the capsid behaves like a nuclear transport receptor to penetrate the nuclear pore, it is fundamentally different. A transport receptor doesn’t need to conceal material for delivery the way the capsid does to avoid detection.
These findings open new lines of inquiry for what the nuclear pore complex is capable of accommodating.
“The HIV-1 capsid is one of the largest things that we now know can go through the nuclear pore complex intact,” Weiskopf says. “It raises all kinds of questions — what other things could be going through the pore that we thought was impossible?”
Schwartz said another question is whether all of the 2,000 nuclear pores in human cells are identical or whether there is something that makes certain pores more amenable to allowing the capsid through.
The capsid is also known to be unusually elastic, a property that may be key for passage through the pore. Another interesting question for the field is whether the cone-shaped capsid gains entry into the pore by squeezing through.
Although the team has shown that the capsid can enter the pore, what happens at the other end of the channel is still unknown — whether the capsid fully or partially enters the nucleus or breaks down inside the channel. Weiskopf is working on perturbing parts of the capsid or the spaghetti-like proteins to learn more about which interactions are most important for successful capsid entry.
Although these results have expanded our understanding of the nuclear pore, much remains unknown, both for HIV-1 infection and for the transport process through the nuclear pore complex.
“The nuclear pore is such an important element of cell biology, we thought it would be interesting to understand it better — and that’s how we figured out that the pore is much bigger than we anticipated,” Schwartz says. “We will certainly try to see whether we can understand the mechanism of HIV-1 infection, how the capsid is released on the other side of the channel, and what factors are important there — and to what extent you can manipulate it or influence it for therapeutic applications.”

Sergey Ovchinnikov uses phylogenetic inference, protein structure prediction/determination, protein design, deep learning, energy-based models, and differentiable programming to tackle evolutionary questions at environmental, organismal, genomic, structural, and molecular scales, with the aim of developing a unified model of protein evolution.