

About 330 miles west of Cambridge lies the small academic town of Ithaca, New York: the location of Cornell University and the hometown of Professor Amy Keating. Surrounded by academics (her father is a professor of computer science at Cornell), Keating was eager to continue her education after high school—just not in Ithaca.
“I could have stayed at Cornell, which is obviously an extremely good school in my hometown, but my family and I agreed that it was important that I go away,” recounts Keating. The scholar/athlete set her sights on Harvard University based on the excellent rowing team and outstanding academics. Physics particularly appealed to her, because it involved using math to explain mechanical and electrical phenomena, and she chose this as her major. She likes to tell people that she also “attempted pure math but failed miserably.” Keating admits that she was not very good at the abstract subject material, and tackling it side-by-side with math whizzes was a harsh awakening after performing well throughout high school. She switched to studying applied math, which was easier for her to manage and also more useful for a physics major.
With an intense rowing schedule, Keating often found herself working late into the night, struggling to solve problems alone. It took a year or two and a serious injury for her to realize that that most of the physics majors were working together in the library many afternoons while she was on the river. “That was very eye-opening. Now I’m a strong advocate of students teaching each other and learning from each other,” explains Keating.
Graduate study gridlock
As she approached the end of her senior year, she had no doubt that she would pursue a PhD, but she did face a crisis about what to study. Initially, she thought she would go to graduate school for physics and applied to and visited many schools. However, she was troubled by the fact that she had tried out a number of areas of physics but never found one that truly captured her interest. In addition to this, Keating began dating a young man, now her husband, who was majoring in chemistry and not set to graduate for another year. “I learned a lot of organic chemistry from him and got very interested in the subject.”
With the decision made to stay in Cambridge for an additional year, she picked up part time work at a Harvard student residence hall cooking, baking, and cleaning in exchange for room and board. Keating also took a few chemistry courses for credit, coached adult rowing, and spent the rest of her time working in the lab of Harvard Physics Professor Mara Prentiss. By the end of that year, she had developed a keen interest in the field of computational chemistry. Having faced difficult decisions about her own post-college plans, she has “a lot of empathy for students who are twenty-one and trying to decide what they want to do in the world.”
Keating and her future husband applied to the same chemistry PhD program at UCLA, where they were both admitted and joined separate labs. She looks back at the interview weekend at UCLA and remembers one faculty interviewer who pointed out the lack of chemistry in her background. “We were talking about cooking, and I told him I like to cook and had been cooking for a job. He said ‘if you can cook, you can do chemistry’, and there is some truth to that, of course.” Keating acknowledges that the first few months of graduate school were traumatic. “I had exactly two undergrad chemistry classes under my belt. I didn’t really know much chemistry and then I was thrown into this PhD program with chemistry majors. And I was taking graduate level courses with my husband, who is a brilliant chemist. But I caught up and managed to learn a lot in a short time.”
Graduate life smoothed out when Keating joined the lab of Ken Houk, a leader in computational physical organic chemistry. Later in her doctoral studies, she added co-advisor Miguel Garcia-Garibay, an expert in experimental photochemistry. Having the two advisors worked out well and led to several joint publications over Keating’s graduate school career. After her husband’s advisor left UCLA for a company, the couple “had to decide what to do. So, we decided we should graduate quickly.” Now married, Keating and her husband earned their PhDs in under five years, but they would continue to be challenged by the “two-body problem” as they formulated a plan for after graduation.
Further afield
The couple knew they both wanted to find postdoc positions, so they looked in cities like San Diego, San Francisco, and Boston, where positions were abundant. Of that time, Keating says: “I was thinking about different problems or fields where my background might apply. I was reading a lot, just to find out what was out there.” This also marks the first time that she started thinking about problems in biology. “I was actually interested in two areas: material science, and biochemistry, both of which are exciting and rapidly growing areas where chemical principles are centrally important.” Keating’s hard work landed her a position back in Cambridge, where she was again co-advised, this time by former MIT Biology Professor Peter Kim at the Whitehead Institute and MIT Professor of Chemistry Bruce Tidor(who was later the founding director of the CSB PhD Program).
The postdoc transition was another time in Keating’s life that she good-naturedly describes as “traumatic,” as she once again had to work to understand all-new vocabulary and experimental methods. Her postdoc provided Keating with her first exposure to large molecules; it was also when she first started working on protein interactions, which would become the crux of her future research. It was in the Kim Lab that she was introduced to coiled-coil proteins. With her background in physics and chemistry, the simplified repeating interactions in these molecules appealed to her. A principle the Keating Lab continues to follow to this day is that they try not to study the most complicated interactions in biology, but rather simpler interactions that they seek to understand in fine detail.
More two-body problems
After four years, Keating hit the academic job market, but she wasn’t sure if she would be accepted as a biochemist because of her change in fields as a postdoc . Her concerns were short-lived as she ended up with a number of exciting offers, including one from MIT. Keating’s husband decided he would go into industry in Boston and with this decision she accepted MIT’s offer to join the Biology faculty in 2002. Later, she added a joint appointment in Biological Engineering.
Keating offers advice to students who are dealing with the two-body problem as she once did.“I think something that helped me and my husband is that we stayed in sync. So, we never had one person make a decision without knowing how that would impact the options of the other person. Of course, that’s not possible for everybody. But that did make our trajectory easier. We would collect our options, put them on the table, look for overlap, and then try to figure out what decision would work best for both of us. And we were very fortunate that we had good options. People have to be flexible to make this work out.” She also recommends looking in cities where there is a high density of opportunities.
The general interest of the Keating lab is in protein-protein interactions, how they work in nature, and how they can be re-engineered using computational and experimental methods. Her group studies proteins that regulate critical processes but are also relatively simple. For example, a system the Keating lab is attracted to is the Bcl-2 family of proteins that control cell death. They have developed a variety of methods that can be used to reprogram the interaction between proteins, and applying these methods to Bcl-2 proteins has generated short peptide molecules that inhibit processes that keep cancer cells alive. Recently the lab has been investigating other types of interactions in cells that are structurally different from the Bcl-2 family. Switching protein families challenges them to develop new methods and allows them to continue to change and evolve their research.
Students and postdocs from the Keating lab have gone on a wide variety of jobs where they study proteins and their interactions in both academia and industry. Keating is happy that young scientists today have so many options. She reflects: “When I was finishing my postdoc, the range of jobs in industry was nothing like it is today. It has been fun to watch my trainees apply their skills to antibody engineering, cancer biology, immuno-oncology and even to start their own companies.” She marvels at how many paths are open to young biologists and likes to tell them that they can’t possibly forsee where they will end up, given the myriad exciting possibilities. Certainly, as a young rower and physics student at Harvard, she had no idea she would end up as a Professor of Biology at MIT.
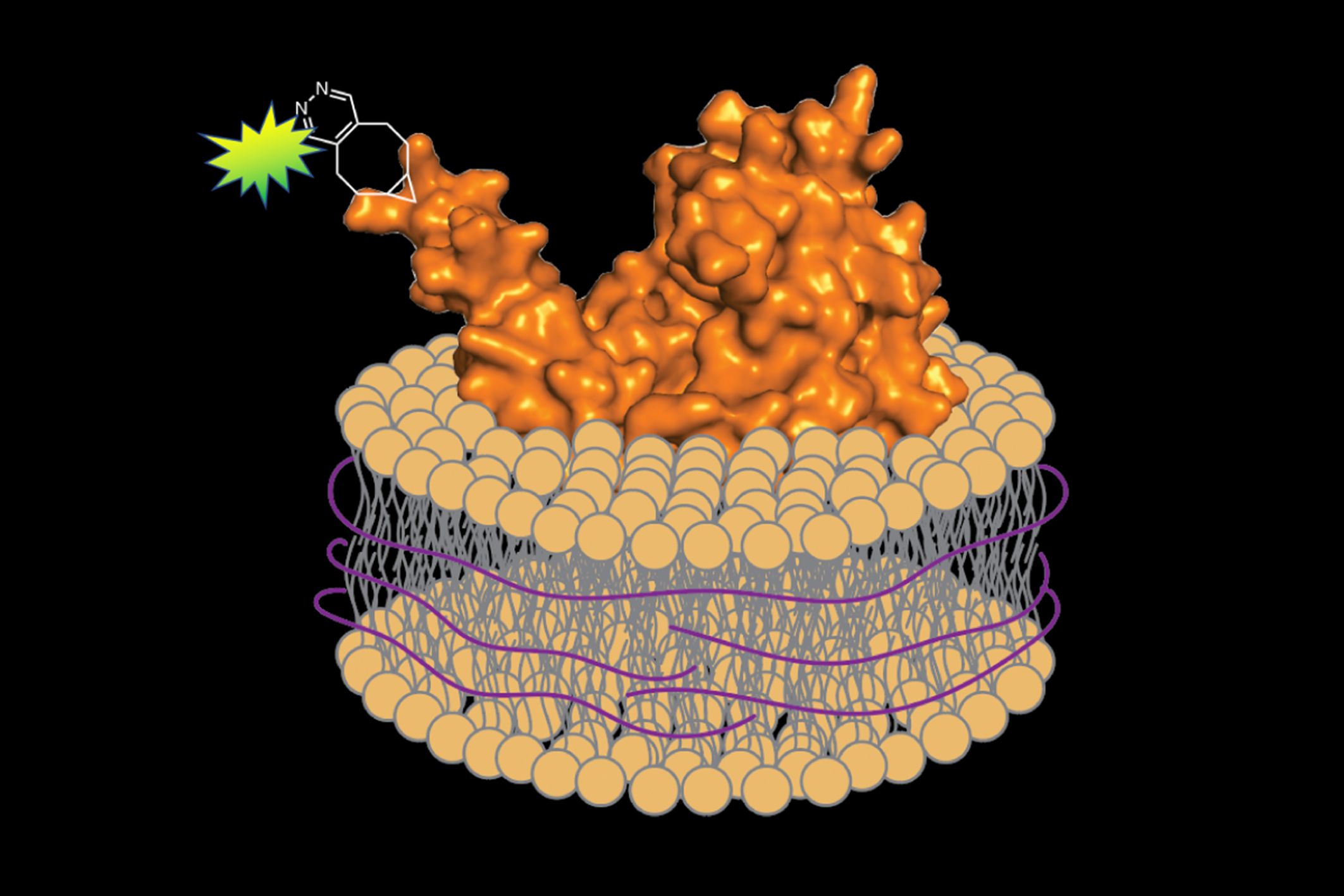
All cells have a lipid membrane that encircles their internal components — forming a protective barrier to control what gets in and what stays out. The proteins embedded in these membranes are essential for life; they help facilitate nutrient transport, energy conversion and storage, and cellular communication. They are also important in human disease, and represent around 60 percent of approved drug targets. In order to study these membrane proteins outside the complexity of the cell, researchers must use detergent to strip away the membrane and extract them. However, determining the best detergent for each protein can involve extensive trial and error. And, removing a protein from its natural environment risks destabilizing the folded structure and disrupting function.
In a study published on Dec. 9 in Cell Chemical Biology, scientists from MIT devised a rapid and generalizable way to extract, purify, and label membrane proteins for imaging without any detergent at all — bringing along a portion of the surrounding membrane to protect the protein and simulate its natural environment. Their approach combines well-established chemical and biochemical techniques in a new way, efficiently isolating the protein so it can be fluorescently labeled and examined under a microscope.
“I always joke that it’s not very lifelike to study proteins in soap,” says senior author Barbara Imperiali, a professor of biology and chemistry. “We’ve created a workflow that allows membrane proteins to be imaged while maintaining their native identities and interactions. Hopefully now fewer people will shy away from studying membrane proteins, given their importance in many physiological processes.”
As a member of the Imperiali lab, former postdoc and lead author Jean-Marie Swiecicki investigated membrane proteins from the foodborne pathogen Campylobacter jejuni. In this study, Swiecicki focused on PglC and PglA, two membrane proteins that play a role in enabling the bacteria to infect human cells. His experiments required labeling PglC and PglA with fluorescent tags in order to track them. However, he wasn’t satisfied with existing methods to do so.
In some cases, the fluorescent tags that must be incorporated into the protein in order to visualize it are too large to be placed at defined positions. In other cases, these tags don’t shine brightly enough, or interfere with the structure and function of the protein.
To avoid such issues, Swiecicki decided to use a method known as “unnatural amino-acid mutagenesis.” Amino acids are the units that compose the protein, and unnatural amino-acid mutagenesis involves adding a new amino acid containing an engineered chemical group within the protein sequence. This chemical group can then be labeled with a brightly glowing tag.
Swiecicki inserted the genetic code for the C. jejuni membrane proteins into a different bacterium, Escherichia coli. Inside E. coli, he could incorporate the unnatural amino acid, which could be chemically modified to add the fluorescent label.
When it came time to remove the proteins from the membrane, he substituted a different substance for the detergent: a polymer of styrene-maleic acid (SMA). Unlike detergent, SMA wraps the extracted protein and a small segment of the associated membrane in a protective shell, preserving its native environment. Imperiali explains, “It’s like a scarf protecting your neck from the cold.”
Swiecicki could then monitor the glowing proteins under a microscope to verify his technique was selective enough to isolate individual membrane proteins. The entire process, he says, takes just a few days, and is generally much faster and more reliable than detergent-based extraction methods, which can take months and require the expertise of highly-trained biochemists to optimize.
“I wouldn’t say it’s a magic bullet that’s going to work for every single protein,” he says. “But it’s a highly efficient tool that could make it easier to study many different kinds of membrane proteins.” Eventually, he says, it may even help facilitate high throughput drug screens.
“As someone who works on membrane protein complexes, I can attest to the great need for better methods to study them,” says Suzanne Walker, a professor of microbiology at Harvard Medical School who was not involved in the study. She hopes to extend the approach outlined in the paper to the protein complexes she investigates in her own lab. “I appreciated the extensive detail included in the text about how to apply the strategy successfully,” she adds.
The next steps will be testing the technique on mammalian proteins, and isolating multiple proteins at once in the SMA shell to observe their interactions. And, of course, every new technique deserves a name. “We’re still working on a catchy acronym,” Imperiali says. “Any ideas?”
This research was funded by the Jane Coffin Childs Memorial Fund for Medical Research, Philippe Foundation, and National Institutes of Health.

Hazel Sive, a globally respected developmental biologist and educator, will become dean of the Northeastern University College of Science, beginning in June 2020. Sive, a member of the Whitehead Institute who has also been on the faculty of the MIT Department of Biology since 1991, is a much-lauded teacher and academic leader at MIT.
“The greatest professional honor of my life has been to be a member of Whitehead Institute and a professor of biology at MIT. To be part of the extraordinary research landscape, to educate our outstanding MIT students, and to have had opportunities to contribute to governance and international activities, has been quite wonderful,” says Sive.
“At the same time, this is an exciting next step in my career,” Sive says. “Northeastern has a fantastic, innovative ethos that meshes with my deep interest in the future of higher education. I look forward to leading the Northeastern College of Science toward even greater excellence in research and education.”
“Hazel has long been committed to teaching and academic leadership, and Northeastern will benefit from her broad experience and expertise,” says David C. Page, Whitehead Institute director and member. “Although we will miss her wit, energy, incisive intelligence, and passionate commitment to outstanding science research, we congratulate her on this wonderful new endeavor.”
Sive, who is also an associate member of Broad Institute of MIT and Harvard, is recognized for her groundbreaking research in vertebrate developmental biology. Her contributions have been wide-ranging, encompassing molecular definition of anterior position, development of the brain ventricular system, and identifying novel cell biological processes, including “epithelial relaxation” and “basal constriction.” Her group defined the extreme anterior domain that gives rise to the mouth and that is a crucial craniofacial signaling center. Sive developed the zebrafish as a tool to analyze human neurodevelopmental disorders, most recently focusing on the metabolic underpinnings of disorders such as autism and 16p11.2 deletion syndrome. She has also been a pioneer in use of the frog Xenopus and zebrafish model systems; indeed, she created the Cold Spring Harbor Laboratory Course on Early Development of Xenopus — which has run for more than 25 years — and she is editor-in-chief of a new two-volume “Xenopus Lab Manual.”
A highly effective educator, Sive has been named a MacVicar Faculty Fellow — MIT’s highest undergraduate teaching accolade — and has twice received the MIT School of Science Teaching Prize. She has taught the undergraduate introductory biology course for 18 years; co-teaches the graduate developmental neuroscience course; and recently created the innovative course Building with Cells for undergraduate and graduate students.
Among her myriad leadership roles, Sive chaired the MIT biology department undergraduate program and has chaired an array of faculty committees. She was the first associate dean of science — where she led the school’s education strategy, promoted diversity in graduate student and faculty recruitment, and devised programs for postdoctoral and junior faculty training. Notably, in 2011 Sive initiated the groundbreaking “Report on the Status of Women Faculty in Science at MIT”.
A native of South Africa — where she earned bachelor’s degrees in chemistry and zoology from the University of Witwatersrand, Johannesburg — Sive has engaged in building connections between MIT and Africa. In 2014, Sive founded the MIT-Africa Initiative, where she serves as faculty director. With the tagline “Collaborating for Impact,” MIT-Africa promotes mutually beneficial engagements in research, education, and innovation. She is founder and faculty director of MISTI-Africa Internships, sending students to multiple African countries. Sive has also focused globally on education, and is founding director of higher education for the MIT Abdul Latif Jameel World Education Lab (J-WEL), located in the Office of Open Learning, started in 2017, that promotes excellence in education across the world. From her leadership, J-WEL Higher Education has built a strong membership across five continents.

Genes passed down from generation to generation play a significant role in determining the traits of every organism. In recent decades, scientists have discovered that another layer of control, known as epigenetics, is also critically important in shaping those characteristics.
Those added controls often work through chemical modifications of genes or other sections of DNA, which influence how easily those genes can be expressed by a cell. Many of those modifications are similar across species, allowing scientists to use plants as an experimental model to uncover how epigenetic processes work.
“Many of the epigenetic phenomena we know about were first discovered in plants, and in terms of understanding the molecular mechanisms, work on plants has also led the way,” says Mary Gehring, an associate professor of biology and a member of MIT’s Whitehead Institute for Biomedical Research.
Gehring’s studies of the small flowering plant Arabidopsis thaliana have revealed many of the mechanisms that underlie epigenetic control, shedding light on how these modifications can be passed from generation to generation.
“We’re trying to understand how epigenetic information is used during plant growth and development, and looking at the dynamics of epigenetic information through development within a single generation, between generations, and on an evolutionary timescale,” she says.
Seeds of discovery
Gehring, who grew up in a rural area of northern Michigan, became interested in plant biology as a student at Williams College, where she had followed her older sister. During her junior year at Williams, she took a class in plant growth and development and ended up working in the lab of the professor who taught the course. There, she studied how development of Arabidopsis is influenced by plant hormones called auxins.
After graduation, Gehring went to work for an environmental consulting company near Washington, but she soon decided that she wanted to go to graduate school to continue studying plant biology. She enrolled at the University of California at Berkeley, where she joined a lab that was studying how different genetic mutations affect the development of seeds.
That lab, led by Robert Fischer, was one of the first to discover an epigenetic phenomenon called gene imprinting in plants. Gene imprinting occurs when an organism expresses only the maternal or paternal version of particular gene. This phenomenon has been seen in flowering plants and mammals.
Gehring’s task was to try to figure out the mechanism behind this phenomenon, focusing on an Arabidopsis imprinted gene called MEDEA. She found that this type of imprinting is achieved by DNA demethylation, a process of removing chemical modifications from the maternal version of the gene, effectively turning it on.
After finishing her PhD in 2005, she worked as a postdoc at the Fred Hutchinson Cancer Research Center, in the lab of Steven Henikoff. There, she began doing larger, genome-scale studies in which she could examine epigenetic markers for many genes at once, instead of one at a time.
During that time, she began studying some of the topics she continues to investigate now, including regulation of the enzymes that control DNA methylation, as well as regulation of “transposable elements.” Also known as “jumping genes,” these sequences of DNA can change their position within the genome, sometimes to promote their own expression at the expense of the organism. Cells often use methylation to silence these genes if they generate harmful mutations.
Patterns of inheritance
After her postdoc, Gehring was drawn to MIT by “how passionate people are about what they’re working on, whether that’s biology or another subject.”
“Boston, especially MIT and Whitehead, is a great environment for science,” she says. “It seemed like there were a lot of opportunities to get really smart and talented students in the lab and have interesting colleagues to talk with.”
When Gehring joined the Whitehead Institute in 2010, she was the only plant biologist on the faculty, but she has since been joined by Associate Professor Jing-Ke Weng.
Her lab now focuses primarily on questions such as how maternal and paternal parents contribute to reproduction, and how their differing interests can lead to genetic conflicts. Gene imprinting is one way that this conflict is played out. Gehring has also discovered that small noncoding RNA molecules play an important role in imprinting and other aspects of inheritance by directing epigenetic modifications such as DNA methylation.
“One thing we’ve found is that this noncoding RNA pathway seems to control the transcriptional dosage of seeds, that is, how many of the transcripts are from the maternally inherited genome and how many from the paternally inherited genome. Not just for imprinted genes, but also more broadly for genes that aren’t imprinted,” Gehring says.
She has also identified a genetic circuit that controls an enzyme that is required to help patterns of DNA methylation get passed from parent to offspring. When this circuit is disrupted, the methylation state changes and unusual traits can appear. In one case, she found that the plants’ leaves become curled after a few generations of disrupted methylation.
“You need this genetic circuit in order to maintain stable methylation patterns. If you don’t, then what you start to see is that the plants develop some phenotypes that get worse over generational time,” she says.
Many of the epigenetic phenomena that Gehring studies in plants are similar to those seen in animals, including humans. Because of those similarities, plant biology has made significant contributions to scientists’ understanding of epigenetics. The phenomenon of epigenomic imprinting was first discovered in plants, in the 1970s, and many other epigenetic phenomena first seen in plants have also been found in mammals, although the molecular details often vary.
“There are a lot of similarities among epigenetic control in flowering plants and mammals, and fungi as well,” Gehring says. “Some of the pathways are plant-specific, like the noncoding RNA pathway that we study, where small noncoding RNAs direct DNA methylation, but small RNAs directing silencing via chromatin is something that happens in many other systems as well.”
Sometimes, unexpected research results are simply due to experimental error. Other times, it’s the opposite — the scientists have uncovered a new phenomenon that reveals an even more accurate portrayal of our bodies and our universe, overturning well-established assumptions. Indeed, many great biological discoveries are made when results defy expectation.
A few years ago, researchers in the Burge lab were comparing the genomic evolution of several different mammals when they noticed a strange pattern. Whenever a new nucleotide sequence appeared in the RNA of one lineage, there was generally an increase in the total amount of RNA produced from the gene in that lineage. Now, in a new paper, the Burge lab finally has an explanation, which redefines our understanding of how genes are expressed.
Once DNA is transcribed into RNA, the RNA transcript must be processed before it can be translated into proteins or go on to serve other roles within the cell. One important component of this processing is splicing, during which certain nucleotide sequences (introns) are removed from the newlymade RNA transcript, while others (the exons) remain. Depending on how the RNA is spliced, a single gene can give rise to a diverse array of transcripts.
Given this order of operations, it makes sense that transcription affects splicing. After all, splicing cannot occur without an RNA transcript. But the inverse theory — that splicing can affect transcription — is now gaining traction. In a recent study, the Burge lab showed that splicing in an exon near the beginning of a gene impacts transcription and increases gene expression, offering an explanation for the patterns in their previous findings.
“Rather than Step A impacting Step B, what we found here is that Step B, splicing, actually feeds back to influence Step A, transcription,” says Christopher Burge, senior author and professor of biology. “It seems contradictory, since splicing requires transcription, but there is actually no contradiction if — as in our model — the splicing of one transcript from a gene influences the transcription of subsequent transcripts from the same gene.”
The study, published on Nov. 28 in Cell, was led by Burge lab postdoc Ana Fiszbein.
Promoting gene expression
In order for transcription to begin, molecular machines must be recruited to a special sequence of DNA, known as the promoter. Some promoters are better at recruiting this machinery than others, and therefore initiate transcription more often. However, having different promoters available to produce slightly different transcripts from a gene helps boost expression and generates transcript diversity, even before splicing occurs mere seconds or minutes later.
At first, Fiszbein wasn’t sure how the new exons were enhancing gene expression, but she theorized that new promoters were involved. Based on evolutionary data available and her experiments at the lab bench, she could see that wherever there was a new exon, there was usually a new promoter nearby. When the exon was spliced in, the new promoter became more active.
The researchers named this phenomenon “exon-mediated activation of transcription starts” (EMATS). They propose a model in which the splicing machinery associated with the new exon recruits transcription machinery to the vicinity, activating transcription from nearby promoters. This process, the researchers predict, likely helps to regulate thousands of mammalian genes across species.
A more flexible genome
Fiszbein believes that EMATS has increased genome complexity over the course of evolution, and may have contributed to species-specific differences. For instance, the mouse and rat genomes are quite similar, but EMATS could have helped produce new promoters, leading to regulatory changes that drive differences in structure and function between the two. EMATS may also contribute to differences in expression between tissues in the same organism.
“EMATS adds a new layer of complexity to gene expression regulation,” Fiszbein says. “It gives the genome more flexibility, and introduces the potential to alter the amount of RNA produced.”
Juan Valcárcel, a research professor at the Catalan Institution for Research and Advanced Studies in the Center for Genomic Regulation in Barcelona, Spain, says understanding the mechanisms behind EMATS could also have biotechnological and therapeutic implications. “A number of human conditions, including genetic diseases and cancer, are caused by a defect or an excess of particular genes,” he says. “Reverting these anomalies through modulation of EMATS might provide innovative therapies.”
Researchers have already begun to tinker with splicing to control transcription. According to Burge, pharmaceutical companies like Ionis, Novartis, and Roche are concocting drugs to regulate splicing and treat diseases like spinal muscular atrophy. There are many ways to decrease gene expression, but it’s much harder to increase it in a targeted manner. “Tweaking splicing might be one way to do that,” he says.
“We found a way in which our cells change gene expression,” Fiszbein adds. “And we can use that to manipulate transcript levels as we want. I think that’s the most exciting part.”
This research was funded by the National Institutes of Health and the Pew Latin American Fellows Program in the Biomedical Sciences.

In order for the instructions contained within a gene to ultimately execute some function in the body, the nucleotides, or letters, that make up the gene’s DNA sequence must be “read” and used to produce a messenger RNA (mRNA). This mRNA must then be translated into a functional protein. A number of different pathways within the cell influence this essential biological process, informing whether, when, and to what extent a gene is expressed. A major class of such regulators are microRNAs (miRNAs). These minute RNAs—they are, on average, 22 nucleotides long—join with a protein called Argonaute to cause certain mRNAs to be degraded, which in turn decreases the amount of translation of those mRNAs into their functional protein forms. Scientists have identified hundreds of miRNAs that are common amongst mammals and other vertebrate animals, and most mammalian mRNAs are targeted by at least one of these miRNAs—an indication of their pervasive importance to our biology. Accurately predicting how any particular miRNA will affect gene expression in a cell is important for understanding our own biology, and might facilitate the design of therapeutic drugs that affect or utilize miRNAs, but the complexity of the miRNA pathway makes this sort of prediction difficult.
The success rate with which a miRNA is able to repress a specific gene (by degrading its mRNA) is called its targeting efficacy, and researchers have used a variety of models to calculate it, with mixed results. In the past, researchers have treated miRNAs as a group and looked at average behavior in order to make predictions, because there simply wasn’t enough data specific to individual miRNAs available to do otherwise. However, Whitehead Institute Member David Bartel, who is also a professor of biology at the Massachusetts Institute of Technology and a Howard Hughes Medical Institute investigator, graduate student Sean McGeary, and former graduate student Kathy Lin collected a massive amount of data on six miRNAs, and from that foundation developed an improved predictive model for all individual miRNAs. Their findings, published online in Science on December 5, provide unprecedented accuracy and granularity in miRNA targeting prediction.
“We used to focus our attention on microRNA targeting patterns that were consistent, because that consistency gave us confidence in what we were seeing,” Bartel says, “but with the robust results of this research, we can now pay attention to differences between individual miRNAs.”
Bartel and the Whitehead Institute Bioinformatics and Research Computing group operate one of the go-to resources for prediction of miRNAs’ targets and target efficacy, known as TargetScan. This latest research will be used to update TargetScan, giving scientists around the world an even more useful reference tool for research involving miRNA-mediated regulation of gene expression.
To understand miRNA targeting, researchers need to identify the particular sites within an mRNA sequence where the miRNA can bind, and they additionally need to know how strong the interaction will be at each site—the binding affinity. In general, a miRNA will bind to an mRNA when there is a match between at least six of the first eight nucleotides of the miRNA and a complementary sequence of nucleotides somewhere on the mRNA. The two sequences are like rows of puzzle pieces being pushed together: if each puzzle piece slots into the corresponding piece, the rows combine into one locked puzzle—the miRNA binds its target. If the pieces don’t fit together, the rows can’t connect. These sorts of binding sites, perfect matches within the first eight nucleotides of the miRNA, are called canonical site types, and researchers used to think that there was a clear hierarchy between them, with each individual site type conferring a similar amount of repression regardless of the miRNA identity. But that’s not what McGeary observed.
McGeary looked at six miRNAs and developed a method to measure, for each miRNA, relative binding affinities to a massive collection of RNA sequences.
“I performed experiments that provide vast numbers of measurements, which collectively inform us on how well a miRNA will bind to an mRNA,” McGeary says.
These measurements, as well as further calculations that McGeary made from them, formed a novel, rich pool of data with which to improve miRNA targeting prediction. From their experiments, the researchers found that the expected targeting hierarchy of canonical sites did not apply to all miRNAs. An individual miRNA might actually have a stronger affinity to one of the canonical sites lower in the expected hierarchy than another. Furthermore, the group discovered that the miRNAs each had unique noncanonical binding sites, some of which were sites that contained at least one mismatch but were still able to bind miRNA. The researchers found many instances in which a miRNA bound more strongly to one of its noncanonical sites than to some of its canonical sites, despite the imperfect or unusual pairing of the noncanonical sites.
“As humans, we like to classify things into discrete buckets with discrete characteristics,” Lin says. “But to build a model that is quantitative, you have to recognize that each miRNA and target interaction is different.”
Factors in a target site’s environment contribute to the individuality of target interactions, as they can affect the structural accessibility of the site for binding. In particular, the researchers found that the four nucleotides closest to a target site could have a huge, even 100-fold combined impact on affinity.
With their high-resolution data, the researchers were able to rigorously verify a supposition within the miRNA research community: that the strength with which a miRNA binds to a target site is the major determinant for how effective that miRNA will be at degrading that mRNA. This striking correlation between site affinity and targeting efficacy also allowed them to create a biochemical model of miRNA targeting that used the vast collection of affinity measurements to predict the efficacy of repression of every mRNA in cell, significantly out-performing all existing models of miRNA targeting. They then used machine learning, in the form of a convolutional neural network developed by Lin, to extend the improved predictions to all miRNAs without the need to generate additional data.
Altogether, these findings paint a much richer picture of miRNA-mediated gene repression. The new level of specificity in miRNA targeting prediction will provide all researchers working on the subject with better information about the impact of a given miRNA in a cell.
This work was supported by the NIH and Howard Hughes Medical Institute.
Written by Greta Friar
***
David Bartel’s primary affiliation is with Whitehead Institute for Biomedical Research, where his laboratory is located and all his research is conducted. He is also a professor of biology at Massachusetts Institute of Technology and investigator with the Howard Hughes Medical Institute.
***
Citation:
“The biochemical basis of microRNA targeting efficacy”
Science, online December 5, 2019, DOI: 10.1126/science.aav1741
Sean E. McGeary (1,2,3†), Kathy S. Lin (1,2,3,4†), Charlie Y. Shi (1,2,3), Thy Pham (1,2,3), Namita Bisaria (1,2,3), Gina M. Kelley (1,2,3), and David P. Bartel (1,2,3,4)
†These authors contributed equally to this work.

Three MIT seniors, Mariam Dogar, Adedoyin Olateru-Olagbegi, and Jessica Quaye, and alumna Jessica Wang ’16, MEng ’17 are recipients of this year’s Schwarzman Scholarship distinguished fellowship. Another alumna was also awarded a scholarship but is waiting to make a public announcement until she has shared the news with her employer.
The five winners were selected from an applicant of pool over 4,700 candidates and will join fellow Schwarzman Scholars from around the world in China next August. Scholars complete a one-year master’s degree in global affairs at Beijing’s Tsinghua University. Their education is complemented by internships, career development mentors, high-profile speakers, and opportunities to travel throughout China.
Inspired by the Rhodes Scholarship, the Schwarzman Scholarship program began in 2015 to bring together talented young leaders and prepare them for the geopolitical and economic challenges of the 21st century by deepening their understanding of China. Since its inception, 18 MIT students and alumni have been named Schwarzman Scholars.
Kim Benard, assistant dean of distinguished fellowships in Career Advising and Professional Development prepares MIT’s applicants, with assistance from the Presidential Committee on Distinguished Fellowships’ faculty members. MIT students and recent alumni interested in learning more about the Schwarzman Scholarship program should contact fellowships@mit.edu.
Hailing from Northborough, Massachusetts, Mariam Dogar is majoring in biology and minoring in urban studies and planning. She aims to make health care more accessible and equitable through reworking outdated policies and utilizing technology. Dogar has worked at the World Bank developing telemedicine policy recommendations for lower middle-income countries. She has two years of experience on the teaching team of MIT’s negotiation and leadership classes, where she shaped pedagogy and co-taught a workshop for MBA students in Malaysia. She has taught humanitarian design in Greece with MIT D-Lab, worked in digital health care investing in the Middle East, and volunteered in refugee programs in Jordan. She is a co-president of MIT Mock Trial and GlobeMed@MIT. She is also an executive member of PaksMIT and counselor for Camp Kesem.
Jessica Quaye, an electrical engineering and computer science major, has conducted research with MIT.nano and the HCIE group in CSAIL. She has also sharpened her technical and business management skills through internships at Google, Microsoft, and Bain and Company. Quaye, a Tau Beta Pi Scholar, is president of the MIT African Students’ Association. She serves on MIT’s Undergraduate Association committees and the EECS Undergraduate Student Advisory Group. She founded the International Students of Color Working Group to support the needs of first-year international students, and she established the first MIT Global Teaching Lab initiative in Ghana. Quaye is from Accra, Ghana. As a Schwarzman Scholar, she hopes to deepen her understanding of public policy and dreams of one day driving policy change in Ghana.
Adedoyin Olateru-Olagbegi, from Hanover, Maryland, is majoring in computer science, economics, and data science. She envisions a world where quality health care is accessible to all, and plans to focus on health in developing countries with an emphasis on innovative digital tools. She has explored her interests in development and public health through classes that have taken her to South Africa and Colombia. As director of Camp Kesem at MIT, Olateru-Olagbegi organizes an annual summer camp for children affected by a parent’s cancer and oversees the MIT students who work with them. She has also held leadership roles with MIT Emergency Medical Services, the MIT Black Students’ Union, and Sigma Kappa Sorority, and has served on several MIT Institute Committees, including as a student advisor to President L. Rafael Reif.
Jessica Wang graduated from MIT in 2016 with a Bachelor of Science in computer science and engineering and received a Master of Engineering in 2017. She is passionate about utilizing technology for good and bringing her joint engineering and design background to shape technology policy. She currently lives in San Francisco, where she builds collaborative design software at Figma. She works on diversity and inclusion initiatives in the workplace and volunteers with Larkin Street, a nonprofit serving homeless youth, as a YCore Fellow. In the past, she’s worked at a machine learning startup, Facebook, and Uber. At MIT, Wang researched online sociopolitical discourse and misinformation, writing her thesis on digital systems to bridge ideological divides. She served as president of MIT Chinese Students’ Club and held leadership positions in MIT TechX and HackMIT.
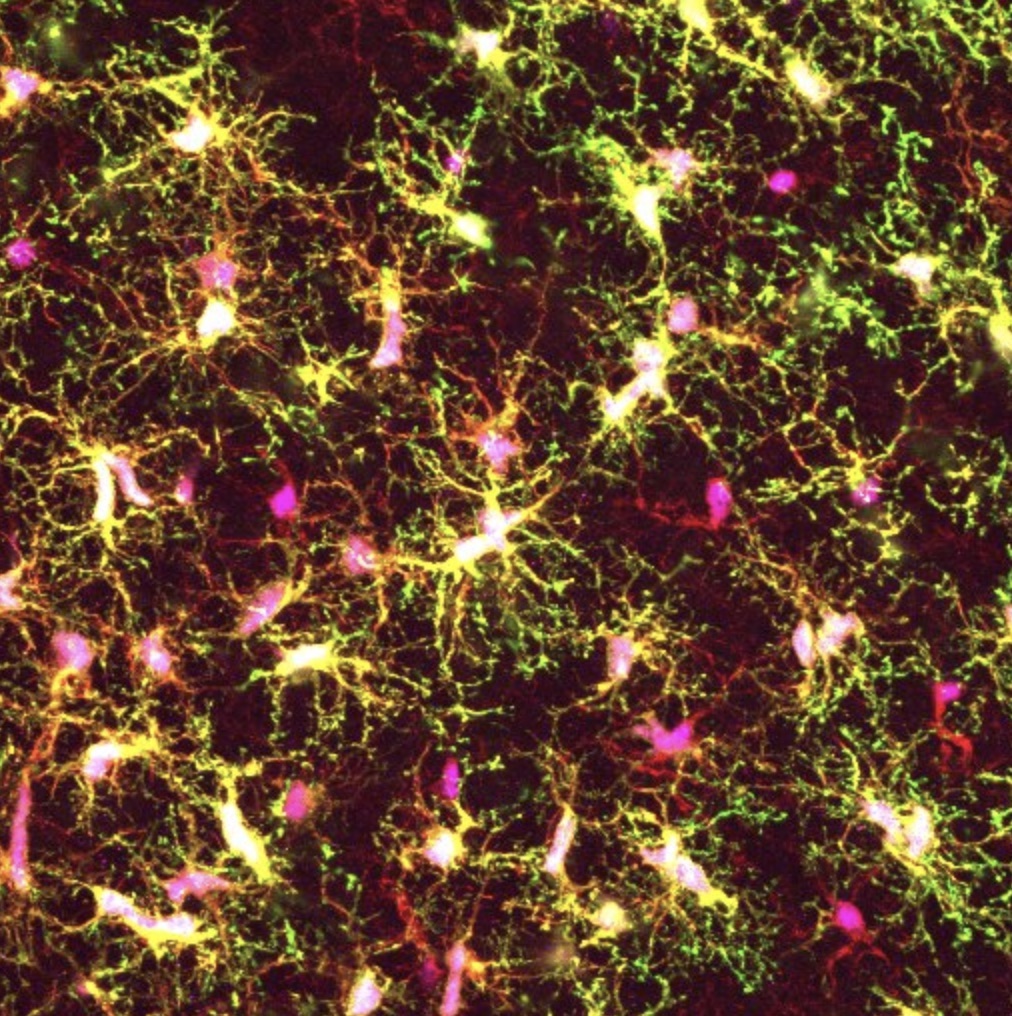
A groundswell of evidence connects defects in the function of microglia, the brain’s resident immune cells, to neurodegenerative diseases, yet the tools for studying these cells in the laboratory have been limited. Now, a team of Whitehead Institute scientists has developed a new experimental platform for generating microglia from human stem cells that includes transplantation into newborn mice. As described online November 26 in the Proceedings of the National Academy of Sciences (PNAS), this new method yields microglial cells that resemble those in the human brain more closely than previous approaches, which could help enable future studies aimed at unravelling the role of microglia in neurodegeneration and other brain disorders.
“The dysfunction of microglia is implicated in a wide variety of brain conditions, and yet our knowledge of them, especially in humans, is really quite limited,” says senior author Rudolph Jaenisch, a Founding Member of the Whitehead Institute and professor of biology at the Massachusetts Institute of Technology. “This new approach will help us lift the hood on these important yet enigmatic brain cells.”
Microglia are increasingly recognized as key players in brain health and disease, but the majority of what is known about them comes from studies of mice, not humans. Yet human and mouse microglia are quite distinct — in humans, the cells are much larger, and have a more branched appearance, suggesting significant differences in their biology.
To address this gap in knowledge, multiple research teams have recently devised methods to generate microglia using human stem cells and grow them under laboratory conditions that mimic their natural environment. However, this approach has a fundamental drawback: the cultured cells do not look like microglia nor do they behave much like them, even though they display the appropriate molecular hallmarks.
“That really suggests to us that this is not the optimal approach to study how microglia are behaving in healthy and diseased brains,” says first author Devon Svoboda, a postdoctoral fellow in the Jaenisch lab. “We set out to create a new method in which the stem-cell derived microglial cells can reside in the brains of mice — one of the best models of the human brain that we have.”
Transplanting human cells into mice — creating “chimeras” — is a well-established technique. However, Svoboda and her colleagues discovered they needed to use special strains of mice that carry human genes for certain growth factors, called cytokines, which are required for microglial development and survival. The researchers utilized mice that carry human genes for four crucial cytokines: CSF1, IL3, SCF, and GM-CSF.
“What is special about these chimeras is really the mice we are using,” says Svoboda. “They express the human alleles of these cytokines which is key because the mouse versions are not able to communicate with receptors on human microglia, so the cells die.”
After transplanting the stem-cell derived microglia into these mice, the research team examined the cells’ morphology and their molecular characteristics. They found that the transplanted cells closely resembled those found in the human brain.
Further analyses revealed some striking differences between the team’s “chimera-grown” microglia and those grown in the laboratory using conventional cell culture methods. Surprisingly, Svoboda and her colleagues found that the cultured microglia showed strong similarities to the diseased microglia from patients with multiple sclerosis, another brain condition in which the cells are implicated.
“If you want to learn more about the role of microglia in disease, then studying them in culture is probably not the best way,” says Svoboda. “The chimeras and the in vitro methods really complement each other, and we think there is a place for both systems in microglia research going forward.”
The Whitehead-led team plans to extend their initial studies in several ways. One is to identify which cytokines and other growth factors are most crucial to microglial development. That knowledge could help improve existing cell culture methods and enable them to more closely mirror the cells’ natural environment. Another key direction is to use the new chimera-based system to create models of neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease, to understand how microglia respond to diseased neurons and, in turn, how diseased microglia can impair neuron function.
“Our chimera-based method will give us a good handle to begin to stringently test the role of microglia in brain health and disease,” says Jaenisch. “This is an important step forward for the field.”
Support for this work was provided by the Cure Alzheimer’s Foundation, MassCATS, and NIH Grants R01 AG058002-01, R01 MH104610, R37 CA084198, and U19 AI131135 (to R.J.). L.D.S. is supported by NIH Grants R24 OD26440, AI32963, and CA034196. J.S. is supported by the National Institute of Child Health and Human Development (K99HD096049).
Written by Nicole Davis
***
Rudolf Jaenisch’s primary affiliation is with Whitehead Institute for Biomedical Research, where his laboratory is located and all his research is conducted. He is also a professor of biology at Massachusetts Institute of Technology.
***
Paper cited:
1. Whitehead Institute, Cambridge, MA 02142, USA
2. Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
3.
4.

Six MIT faculty members have been elected as fellows of the American Association for the Advancement of Science (AAAS).
The new fellows are among a group of 443 AAAS members elected by their peers in recognition of their scientifically or socially distinguished efforts to advance science. This year’s fellows will be honored at a ceremony on Feb. 15, at the AAAS Annual Meeting in Seattle.
Arthur B. Baggeroer is a professor of mechanical, ocean and electrical engineering, the Ford Professor of Engineering, Emeritus, and an international authority on underwater acoustics. Throughout his career he made significant advances to geophysical signal processing and sonar technology, in addition to serving as a long-time intellectual resource to the U.S. Navy.
Suzanne Flynn is a professor of linguistics and language acquisition, and a leading researcher on the acquisition of various aspects of syntax by children and adults in bilingual, second- and third-language contexts. She also works on the neural representation of the multilingual brain and issues related to language impairment, autism, and aging. Flynn is currently editor-in-chief and a co-founding editor of Syntax: A Journal of Theoretical, Experimental and Interdisciplinary Research.
Wesley L. Harris is the Charles Stark Draper Professor of Aeronautics and Astronautics and has served as MIT associate provost and head of the Department of Aeronautics and Astronautics. His academic research program includes unsteady aerodynamics, aeroacoustics, rarefied gas dynamics, sustainment of capital assets, and chaos in sickle cell disease. Prior to coming to MIT, he was a NASA associate administrator, responsible for all programs, facilities, and personnel in aeronautics.
Eric Klopfer is a professor and head of the Comparative Media Studies/Writing program and the director of the Scheller Teacher Education Program and The Education Arcade at MIT. His interests range from the design and development of new technologies for learning to professional development and implementation in schools. Much of Klopfer’s research has focused on computer games and simulations for building understanding of science, technology, engineering, and mathematics.
Douglas Lauffenburger, is the Ford Professor of Biological Engineering, Chemical Engineering, and Biology, and head of the Department of Biological Engineering. He and his research group investigate the interface of bioengineering, quantitative cell biology, and systems biology. The lab’s main focus has been on fundamental aspects of cell dysregulation, complemented by translational efforts in identifying and testing new therapeutic ideas.
John J. Leonard is the Samuel C. Collins Professor of Mechanical and Ocean Engineering and a leading expert in navigation and mapping for autonomous mobile robots. His research focuses on long-term visual simultaneous localization and mapping in dynamic environments. In addition to underwater vehicles, Leonard has applied his pursuit of persistent autonomy to the development of self-driving cars.
This year’s fellows will be formally announced in the AAAS News and Notes section of Science on Nov. 28.