Whitehead Institute and CSAIL researchers created a machine-learning model to predict and generate protein localization, with implications for understanding and remedying disease.
Greta Friar | Whitehead Institute
Proteins are the workhorses that keep our cells running, and there are many thousands of types of proteins in our cells, each performing a specialized function. Researchers have long known that the structure of a protein determines what it can do. More recently, researchers are coming to appreciate that a protein’s localization is also critical for its function. Cells are full of compartments that help to organize their many denizens. Along with the well-known organelles that adorn the pages of biology textbooks, these spaces also include a variety of dynamic, membrane-less compartments that concentrate certain molecules together to perform shared functions. Knowing where a given protein localizes, and who it co-localizes with, can therefore be useful for better understanding that protein and its role in the healthy or diseased cell, but researchers have lacked a systematic way to predict this information.
Meanwhile, protein structure has been studied for over half-a-century, culminating in the artificial intelligence tool AlphaFold, which can predict protein structure from a protein’s amino acid code, the linear string of building blocks within it that folds to create its structure. AlphaFold and models like it have become widely used tools in research.
Proteins also contain regions of amino acids that do not fold into a fixed structure, but are instead important for helping proteins join dynamic compartments in the cell. MIT Professor Richard Young and colleagues wondered whether the code in those regions could be used to predict protein localization in the same way that other regions are used to predict structure. Other researchers have discovered some protein sequences that code for protein localization, and some have begun developing predictive models for protein localization. However, researchers did not know whether a protein’s localization to any dynamic compartment could be predicted based on its sequence, nor did they have a comparable tool to AlphaFold for predicting localization.
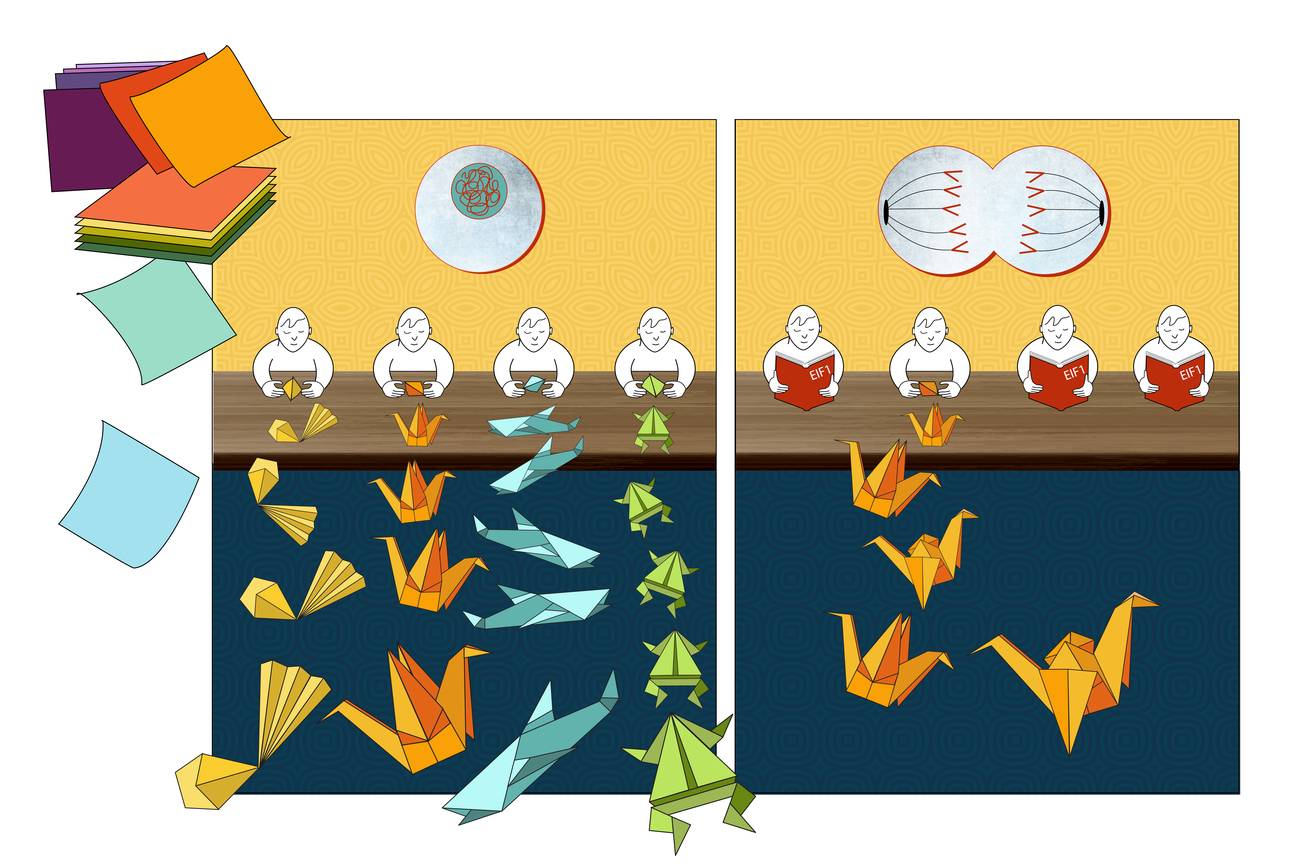
Now, Young, also member of the Whitehead Institute for Biological Research; Young lab postdoc Henry Kilgore; Regina Barzilay, the School of Engineering Distinguished Professor for AI and Health at MIT’s Computer Science and Artificial Intelligence Laboratory (CSAIL); and colleagues have built such a model, which they call ProtGPS. In a paper published on Feb. 6 in the journal Science, with first authors Kilgore and Barzilay lab graduate students Itamar Chinn, Peter Mikhael, and Ilan Mitnikov, the cross-disciplinary team debuts their model. The researchers show that ProtGPS can predict to which of 12 known types of compartments a protein will localize, as well as whether a disease-associated mutation will change that localization. Additionally, the research team developed a generative algorithm that can design novel proteins to localize to specific compartments.
“My hope is that this is a first step towards a powerful platform that enables people studying proteins to do their research,” Young says, “and that it helps us understand how humans develop into the complex organisms that they are, how mutations disrupt those natural processes, and how to generate therapeutic hypotheses and design drugs to treat dysfunction in a cell.”
The researchers also validated many of the model’s predictions with experimental tests in cells.
“It really excited me to be able to go from computational design all the way to trying these things in the lab,” Barzilay says. “There are a lot of exciting papers in this area of AI, but 99.9 percent of those never get tested in real systems. Thanks to our collaboration with the Young lab, we were able to test, and really learn how well our algorithm is doing.”
Developing the model
The researchers trained and tested ProtGPS on two batches of proteins with known localizations. They found that it could correctly predict where proteins end up with high accuracy. The researchers also tested how well ProtGPS could predict changes in protein localization based on disease-associated mutations within a protein. Many mutations — changes to the sequence for a gene and its corresponding protein — have been found to contribute to or cause disease based on association studies, but the ways in which the mutations lead to disease symptoms remain unknown.
Figuring out the mechanism for how a mutation contributes to disease is important because then researchers can develop therapies to fix that mechanism, preventing or treating the disease. Young and colleagues suspected that many disease-associated mutations might contribute to disease by changing protein localization. For example, a mutation could make a protein unable to join a compartment containing essential partners.
They tested this hypothesis by feeding ProtGOS more than 200,000 proteins with disease-associated mutations, and then asking it to both predict where those mutated proteins would localize and measure how much its prediction changed for a given protein from the normal to the mutated version. A large shift in the prediction indicates a likely change in localization.
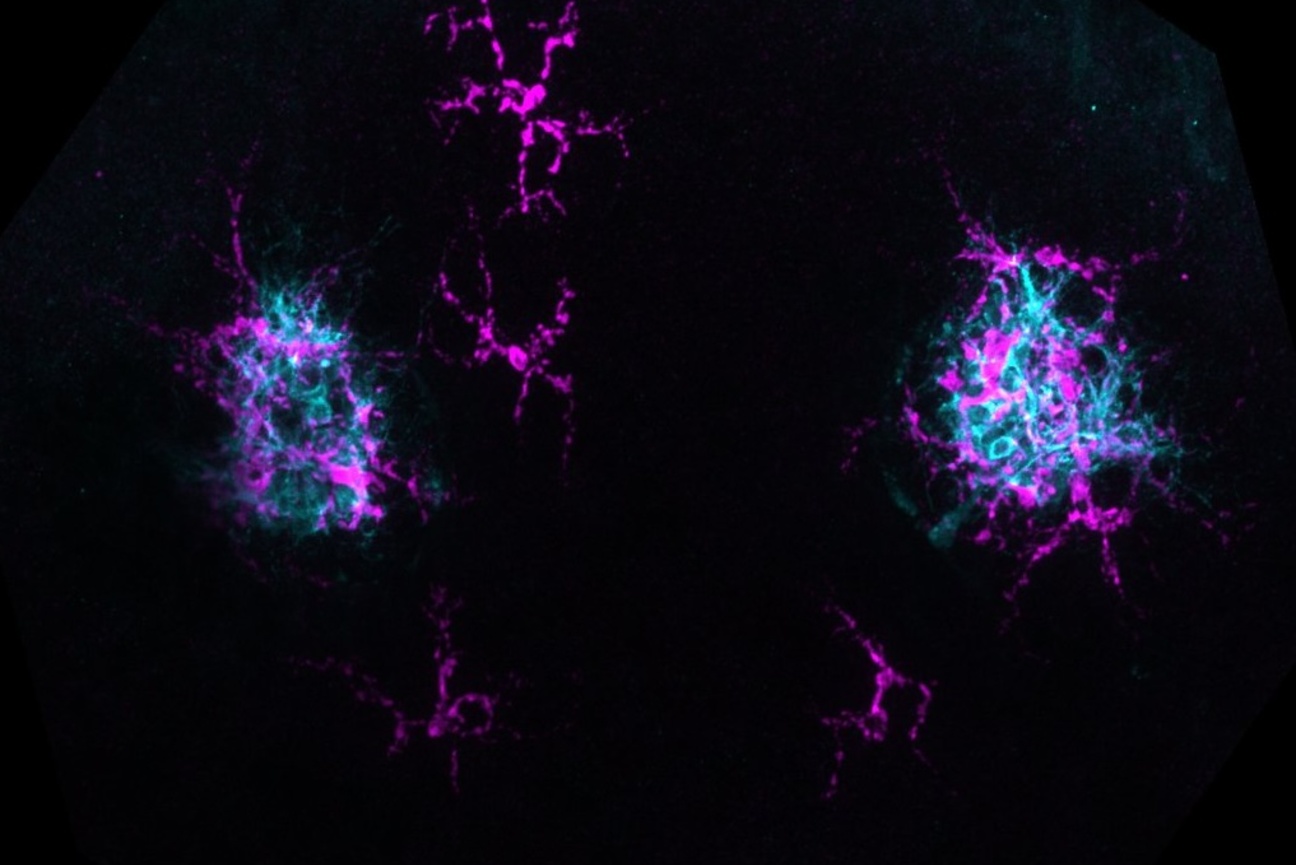
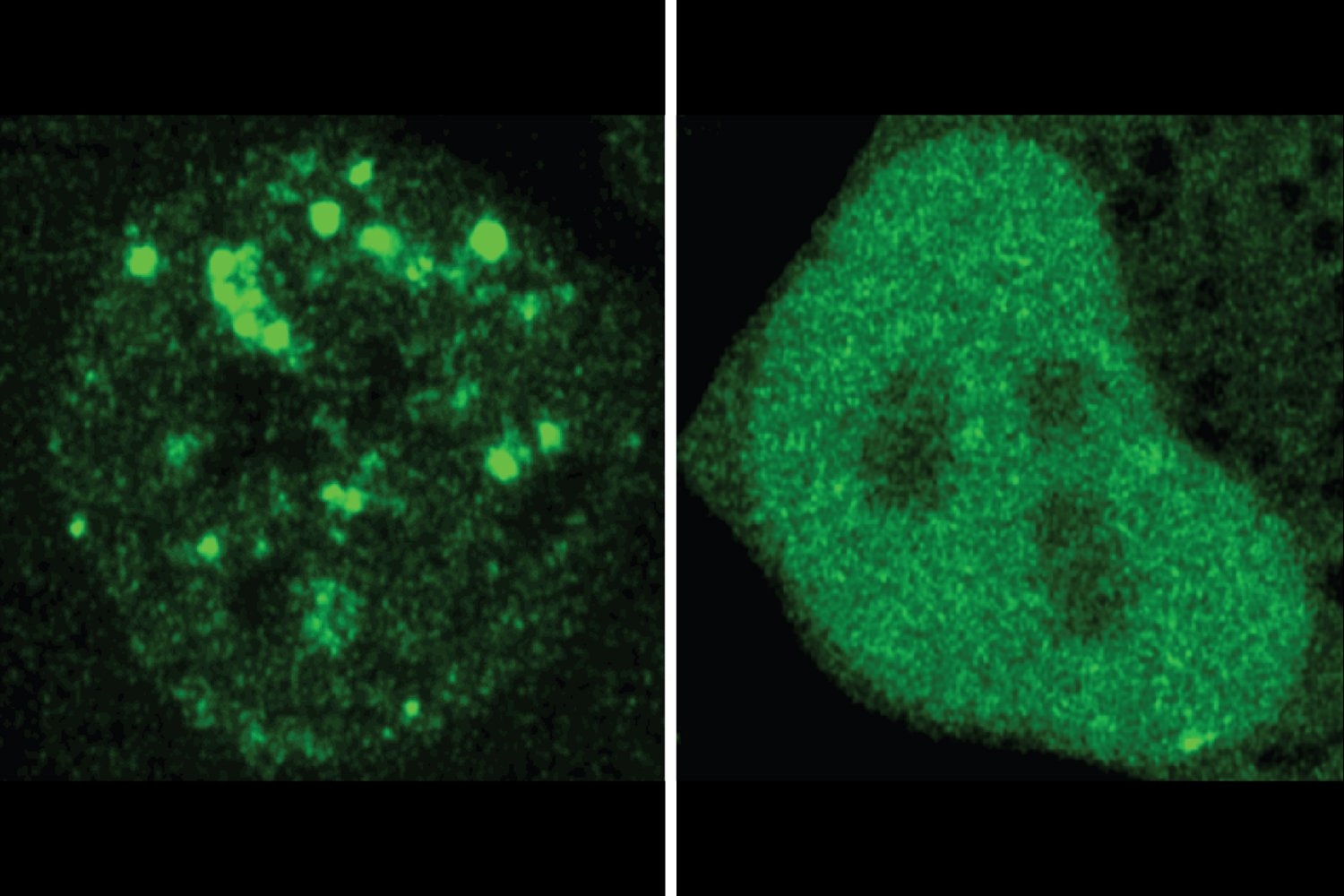
The researchers found many cases in which a disease-associated mutation appeared to change a protein’s localization. They tested 20 examples in cells, using fluorescence to compare where in the cell a normal protein and the mutated version of it ended up. The experiments confirmed ProtGPS’s predictions. Altogether, the findings support the researchers’ suspicion that mis-localization may be an underappreciated mechanism of disease, and demonstrate the value of ProtGPS as a tool for understanding disease and identifying new therapeutic avenues.
“The cell is such a complicated system, with so many components and complex networks of interactions,” Mitnikov says. “It’s super interesting to think that with this approach, we can perturb the system, see the outcome of that, and so drive discovery of mechanisms in the cell, or even develop therapeutics based on that.”
The researchers hope that others begin using ProtGPS in the same way that they use predictive structural models like AlphaFold, advancing various projects on protein function, dysfunction, and disease.
Moving beyond prediction to novel generation
The researchers were excited about the possible uses of their prediction model, but they also wanted their model to go beyond predicting localizations of existing proteins, and allow them to design completely new proteins. The goal was for the model to make up entirely new amino acid sequences that, when formed in a cell, would localize to a desired location. Generating a novel protein that can actually accomplish a function — in this case, the function of localizing to a specific cellular compartment — is incredibly difficult. In order to improve their model’s chances of success, the researchers constrained their algorithm to only design proteins like those found in nature. This is an approach commonly used in drug design, for logical reasons; nature has had billions of years to figure out which protein sequences work well and which do not.
Because of the collaboration with the Young lab, the machine learning team was able to test whether their protein generator worked. The model had good results. In one round, it generated 10 proteins intended to localize to the nucleolus. When the researchers tested these proteins in the cell, they found that four of them strongly localized to the nucleolus, and others may have had slight biases toward that location as well.
“The collaboration between our labs has been so generative for all of us,” Mikhael says. “We’ve learned how to speak each other’s languages, in our case learned a lot about how cells work, and by having the chance to experimentally test our model, we’ve been able to figure out what we need to do to actually make the model work, and then make it work better.”
Being able to generate functional proteins in this way could improve researchers’ ability to develop therapies. For example, if a drug must interact with a target that localizes within a certain compartment, then researchers could use this model to design a drug to also localize there. This should make the drug more effective and decrease side effects, since the drug will spend more time engaging with its target and less time interacting with other molecules, causing off-target effects.
The machine learning team members are enthused about the prospect of using what they have learned from this collaboration to design novel proteins with other functions beyond localization, which would expand the possibilities for therapeutic design and other applications.
“A lot of papers show they can design a protein that can be expressed in a cell, but not that the protein has a particular function,” Chinn says. “We actually had functional protein design, and a relatively huge success rate compared to other generative models. That’s really exciting to us, and something we would like to build on.”
All of the researchers involved see ProtGPS as an exciting beginning. They anticipate that their tool will be used to learn more about the roles of localization in protein function and mis-localization in disease. In addition, they are interested in expanding the model’s localization predictions to include more types of compartments, testing more therapeutic hypotheses, and designing increasingly functional proteins for therapies or other applications.
“Now that we know that this protein code for localization exists, and that machine learning models can make sense of that code and even create functional proteins using its logic, that opens up the door for so many potential studies and applications,” Kilgore says.