Researchers at the Whitehead Institute have described a pathyway by which planarians, freshwater flatworms with spectacular regenerative capabilities, can restore large portions of their nervous system, even regenerating a new head with a fully functional brain.
Shafaq Zia | Whitehead Institute
February 6, 2025
Cut off any part of this worm’s body and it will regrow. This is the spectacular yet mysterious regenerative ability of freshwater flatworms known as planarians. The lab of Whitehead Institute Member Peter Reddien investigates the principles underlying this remarkable feat. In their latest study, published in PLOS Genetics on February 6, first author staff scientist M. Lucila Scimone, Reddien, and colleagues describe how planarians restore large portions of their nervous system—even regenerating a new head with a fully functional brain—by manipulating a signaling pathway.
This pathway, called the Delta-Notch signaling pathway, enables neurons to guide the differentiation of a class of progenitors—immature cells that will differentiate into specialized types—into glia, the non-neuronal cells that support and protect neurons. The mechanism ensures that the spatial pattern and relative numbers of neurons and glia at a given location are precisely restored following injury.
“This process allows planarians to regenerate neural circuits more efficiently because glial cells form only where needed, rather than being produced broadly within the body and later eliminated,” said Reddien, who is also a professor of biology at Massachusetts Institute of Technology and an Investigator with the Howard Hughes Medical Institute.
Coordinating regeneration
Multiple cell types work together to form a functional human brain. These include neurons and a more abundant group of cells called glial cells—astrocytes, microglia, and oligodendrocytes. Although glial cells are not the fundamental units of the nervous system, they perform critical functions in maintaining the connections between neurons, called synapses, clearing away dead cells and other debris, and regulating neurotransmitter levels, effectively holding the nervous system together like glue. A few years ago, Reddien and colleagues discovered cells in planarians that looked like glial cells and performed similar neuro-supportive functions. This led to the first characterization of glial cells in planarians in 2016.
Unlike in mammals where the same set of neural progenitors give rise to both neurons and glia, glial cells in planarians originate from a separate, specialized group of progenitors. These progenitors, called phagocytic progenitors, can not only give rise to glial cells but also pigment cells that determine the worm’s coloration, as well as other, lesser understood cell types.
Why neurons and glia in planarians originate from distinct progenitors—and what factors ultimately determine the differentiation of phagocytic progenitors into glia—are questions that still puzzled Reddien and team members. Then, a study showing that planarian neurons regenerate before glia formation led the researchers to wonder whether a signaling mechanism between neurons and phagocytic progenitors guides the specification of glia in planarians.
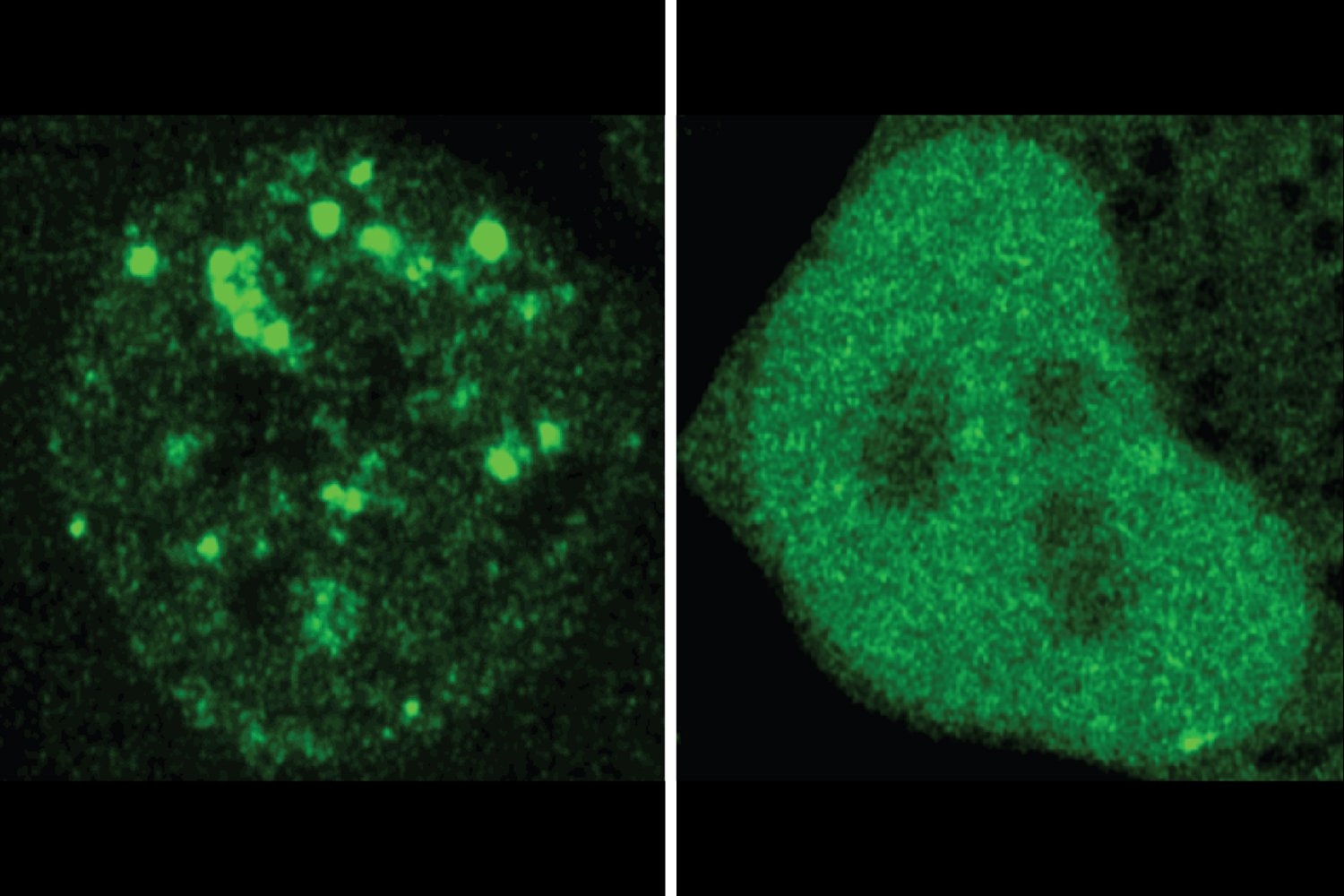
The first step to unravel this mystery was to look at the Notch signaling pathway, which is known to play a crucial role in the development of neurons and glia in other organisms, and determine its role in planarian glia regeneration. To do this, the researchers used RNA interference (RNAi)—a technique that decreases or completely silences the expression of genes—to turn off key genes involved in the Notch pathway and amputated the planarian’s head. It turned out Notch signaling is essential for glia regeneration and maintenance in planarians—no glial cells were found in the animal following RNAi, while the differentiation of other types of phagocytic cells was unaffected.
Of the different Notch signaling pathway components the researchers tested, turning of the genes notch-1, delta-2, and suppressor of hairless produced this phenotype. Interestingly, the signaling molecules Delta-2 was found on the surface of neurons, whereas Notch-1 was expressed in phagocytic progenitors.
With these findings in hand, the researchers hypothesized that interaction between Delta-2 on neurons and Notch-1 on phagocytic progenitors could be governing the final fate determination of glial cells in planarians.
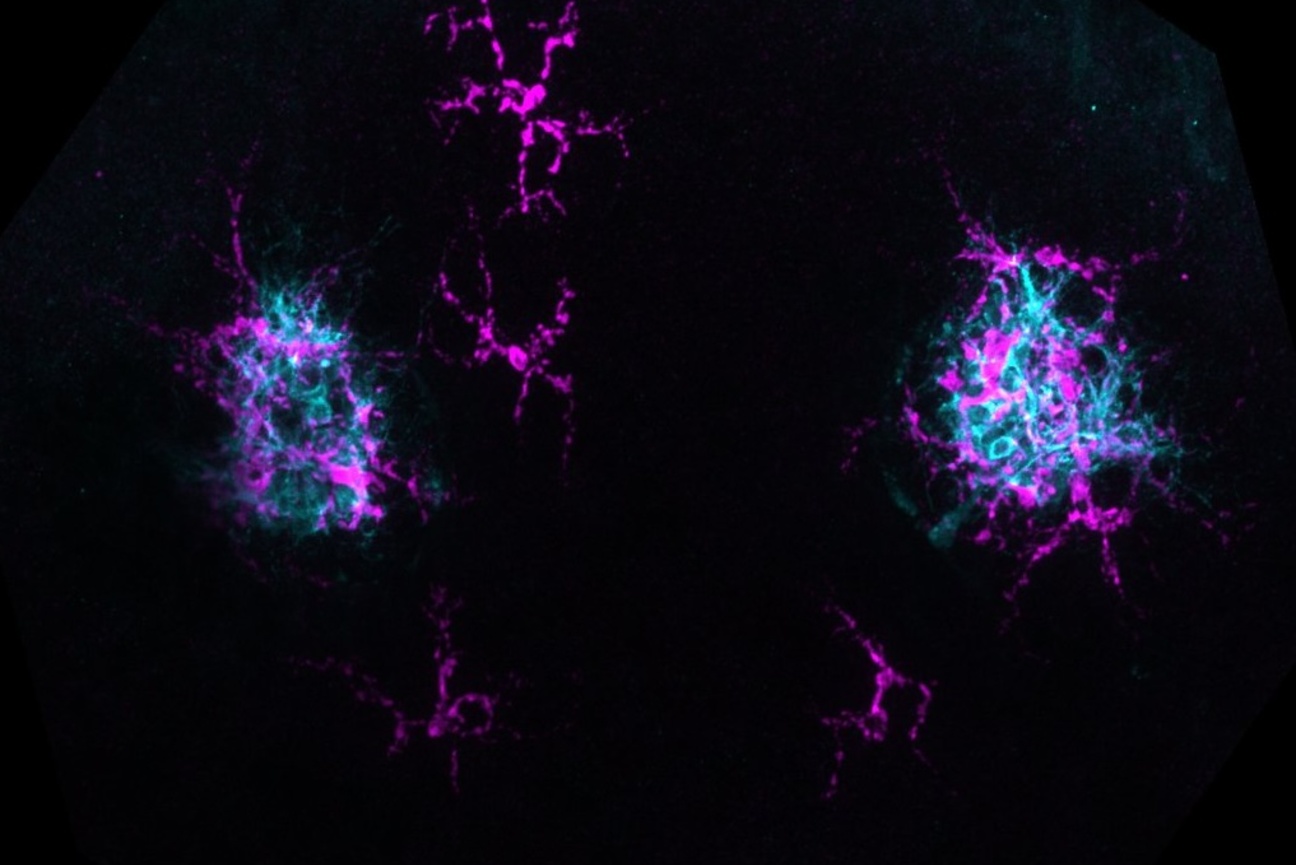
To test the hypothesis, the researchers transplanted eyes either from planarians lacking the notch-1 gene or from planarians lacking the delta-2 gene into wild-type animals and assessed the formation of glial cells around the transplant site. They observed that glial cells still formed around the notch-1 deficient eyes, as notch-1 was still active in the glial progenitors of the host wild-type animal. However, no glial cells formed around the delta-2 deficient eyes, even with the Notch signaling pathway intact in phagocytic progenitors, confirming that delta-2 in the photoreceptor neurons is required for the differentiation of phagocytic progenitors into glia near the eye.
“This experiment really showed us that you have two faces of the same coin—one is the phagocytic progenitors expressing Notch-1, and one is the neurons expressing Delta-2—working together to guide the specification of glia in the organism,”said Scimone.
The researchers have named this phenomenon coordinated regeneration, as it allows neurons to influence the pattern and number of glia at specific locations without the need for a separate mechanism to adjust the relative numbers of neurons and glia.
The group is now interested in investigating whether the same phenomenon might also be involved in the regeneration of other tissue types.