Nicole Davis | Whitehead Institute
November 27, 2018
Cambridge, MA. – The ability to regrow missing or damaged body parts is one of the great marvels of modern biology. In an effort to lay bare the biological underpinnings of this phenomenon, scientists at Whitehead Institute have begun to define the core features that are required for regeneration in flatworms. Their research, which appears online November 27 in Cell Reports, reveals that a set of cellular and molecular responses — previously thought to be essential for regeneration following amputations and other major injuries — is in fact dispensable.
“This is a real surprise,” said senior author Peter Reddien, a Member of Whitehead Institute, professor of biology at Massachusetts Institute of Technology, and investigator with the Howard Hughes Medical Institute. “These responses are broad, prominent attributes of tissue regeneration and repair and, a reasonable bet was that they function to bring about regeneration.”
About eight years ago, Reddien and his team described a set of biological activities that are triggered by injuries that remove tissue. Whereas a cut or a scrape removes little if any tissue, more damaging injuries, like amputations, cause significant tissue loss. That missing tissue must be regenerated to ensure the organism retains its proper anatomical proportions.
A series of cellular and molecular activities — known collectively as the missing tissue response — were believed to enable this regeneration to occur. They include the sustained action of genes that respond to injury, a period of intense cell division in areas surrounding the wound, and a general increase in cell death throughout the body. “This happens prominently, not only in planarians but also in other organisms capable of regeneration, so we suspected that the missing tissue response must play a very fundamental role in regeneration,” recalled Reddien.
What types of injuries require the missing tissue response for repair, and what is the function of the missing tissue response in regeneration? Graduate student and first author Aneesha Tewari, Reddien and colleagues, including Sarah Stern and Isaac Oderberg, set out to uncover the answers. This work forms the basis of their latest Cell Reports study.
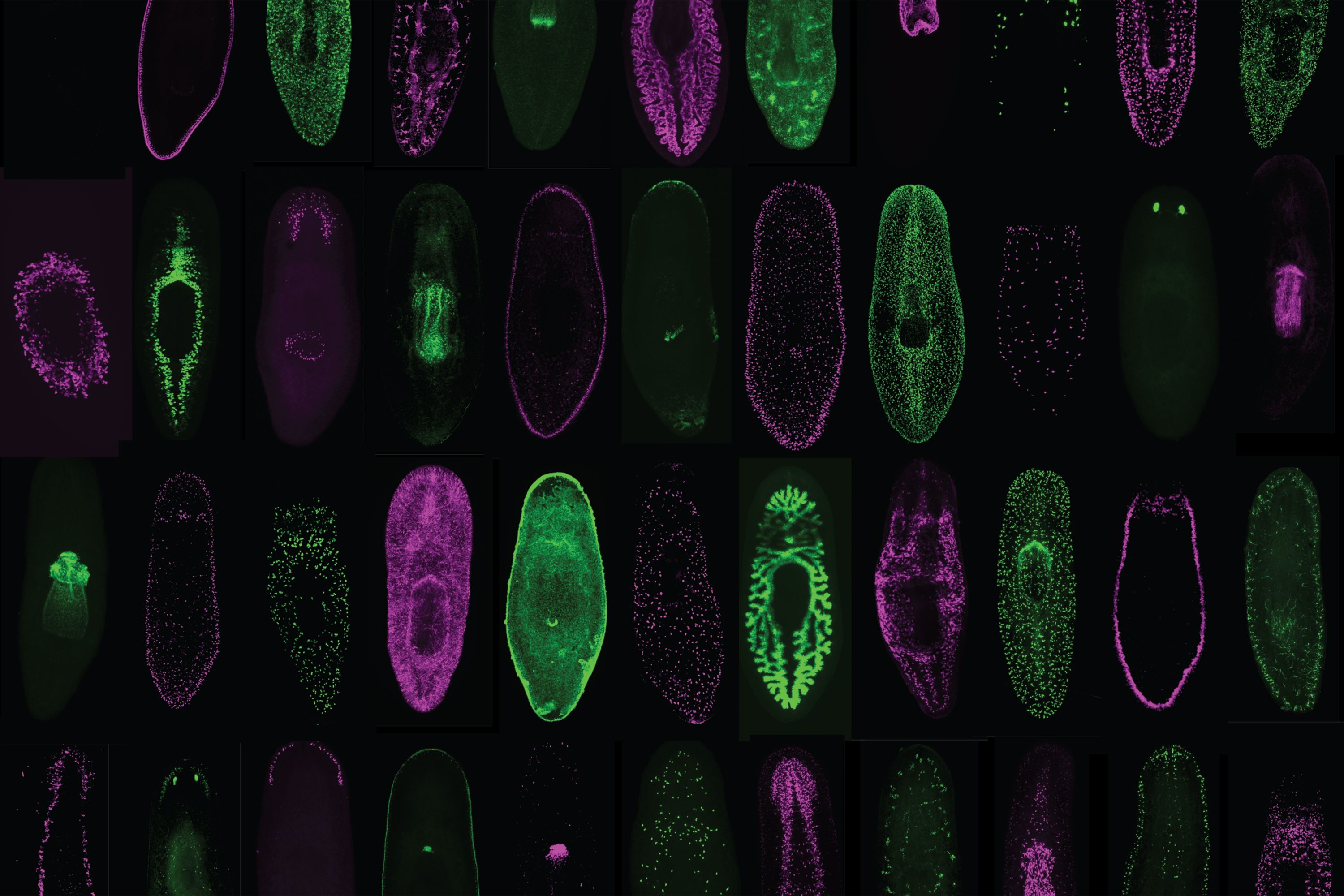
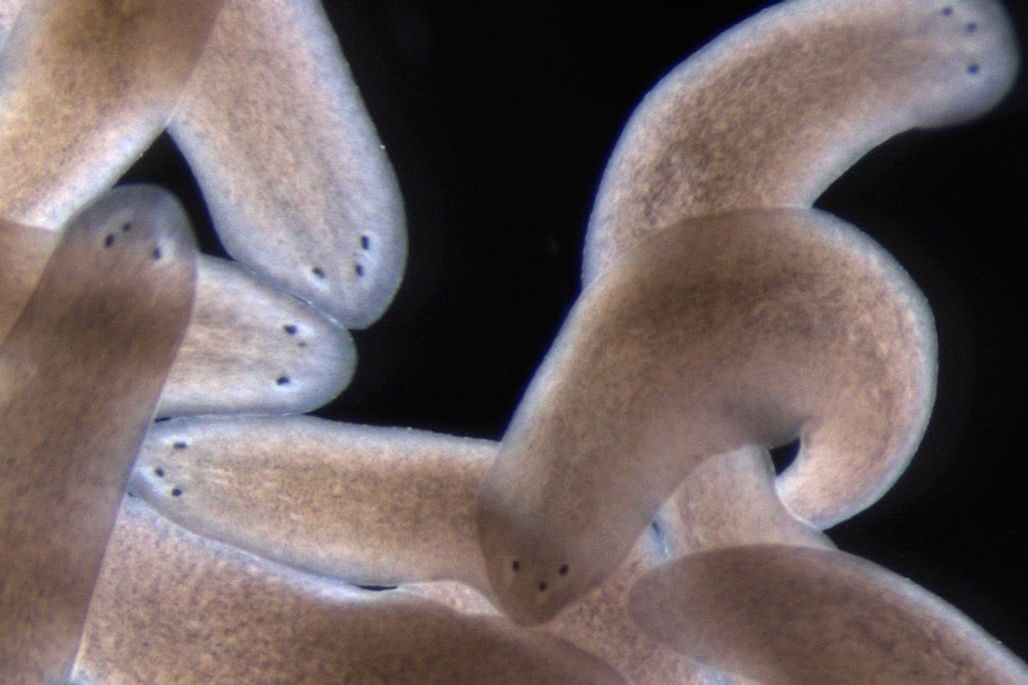
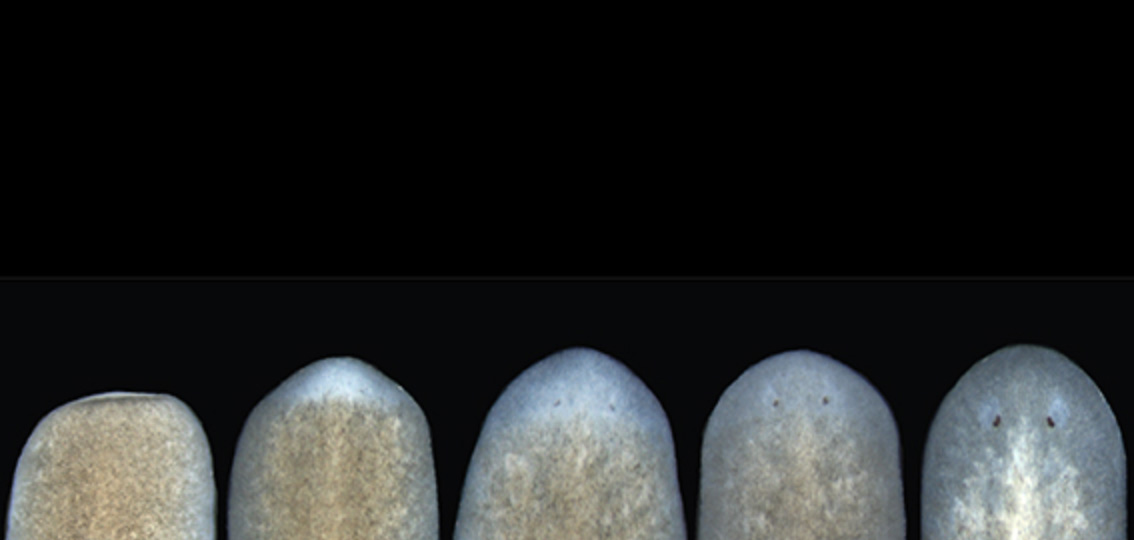
The researchers harnessed an earlier discovery that a gene known as follistatin is required for the missing tissue response in flatworms (known as planarians). By using molecular tools to inhibit this gene, they could block the missing tissue response and observe what happens under various wound conditions, ranging from minimal (the removal of an eye, for example) to moderate (the removal of the pharynx or part of the head) to significant tissue loss (the removal of a complete side of the body). Remarkably, in every case, the missing tissue was regenerated, albeit much more slowly than it would be otherwise.
“These results tell us that what the missing tissue response is really doing is simply pushing the foot down on the gas pedal — basically accelerating the process of regeneration,” explained Reddien. “If you can’t accelerate, you’ll still get there, it just takes longer.”
Tewari, Reddien, and their colleagues also cracked a thorny mystery surrounding the missing tissue response. Although their results show that it is not required across a wide range of injuries, there is one lingering instance in which regeneration failed to occur when they blocked the missing tissue response: head amputation.
“This was a big puzzle,” said Tewari. “It left us wondering whether or not we could generalize our findings to all types of wounds — is there something special about the head that makes it uniquely dependent on the missing tissue response?”
The answer, it turns out, is no. When follistatin is blocked, a key signaling protein, called Wnt1, kicks into overdrive. And when that happens, the tissue destined to form the head does not receive the positional cues it needs to properly regrow, which means regeneration fails to proceed. But, when both the missing tissue response and Wnt1 are blocked, the head does indeed regenerate, the team uncovered.
Taken together, the researchers’ findings begin to clarify what is essential for regeneration to take place and what is not. “Our study greatly simplifies the picture of what it takes to regenerate,” said Reddien. “And that’s an important step along the path towards dissecting the central elements of regeneration in animals that do regenerate well, like flatworms, and then applying that knowledge to understand what the limits might be in those animals that don’t regenerate as well, like humans.”
This research was supported by the NIH (R01GM080639), the National Science Foundation, the Eleanor Schwartz Charitable Foundation, and the Howard Hughes Medical Institute.