To deconstruct and perhaps demystify discovery, let’s unwrap the inside story of a paper published by the lab of Troy Littleton, Menicon Professor of Neuroscience in the Departments of Biology and Brain and Cognitive Sciences. The study reported important findings both about a possible mechanism of seizures in epilepsy, which affects 60 million people worldwide, and also the underappreciated relationship between neurons and the brain cells called glia that help them function. Through four years of work led by former postdoc Shirley Weiss, Littleton’s team thoroughly unraveled the complex breakdown that makes fruit flies with a genetic mutation prone to seizures and showed multiple ways to intervene, including with human medicines. Published in April 2019 in eLife, a far-reaching “open access” journal that is free for all to read, the paper has since been viewed thousands of times.
Figuring out the puzzle of exactly how the mutation made glial cells fail to prevent seizures was a source of particular excitement for Weiss. Littleton adds that the discovery could open up new strategies for developing drugs to address epilepsy in humans, which the flies model well.
“One of the long-term motivations for the field in general, not just our lab, is there might be pharmacological access to glial cells that might have less side effects than would happen if you target neurons directly,” he said.
Too much excitement
First, a little biological background. Neurons are electrical. Their participation in the brain circuits that guide behavior, emotion, reasoning and memory depends on how they build up or dissipate electrical charge by taking in or ejecting ions of calcium, potassium and sodium. If they remain too electrically charged up because of an imbalance of these ions, they can become hyperactive and, in groups, produce seizures. In this study, it turned out that a certain kind of glial cells were responsible for regulating the balance of potassium ions around their neuronal neighbors to help govern their electrical charge and activity. The mutation, Weiss discovered, caused the glia to leave too much potassium outside the neurons, making it harder for the neurons to get rid of the potassium they had built up inside when they were electrically active. Without the ability to get their potassium out, the neurons stayed too excited, producing seizures.
The lab first discovered the mutation, which the Louisianan Littleton named “zydeco,” in 2005 when a team led by Zhuo Guan used genetic screening techniques Littleton learned when he was a postdoc in the 1990s at the University of Wisconsin. The team’s broader goal was to learn about how neurons communicate with each other, so they looked for flies with mutations that either shut the process down, leading to a readily observable symptom of paralysis, or amped it way up, leading to seizures. Zydeco fell into that second category, making fruit flies seize dramatically when stressed by heat or by getting jostled around.
“It was so striking, it was hard to ignore,” Littleton said. “Whatever this gene was, it was doing something very important in the brain.”
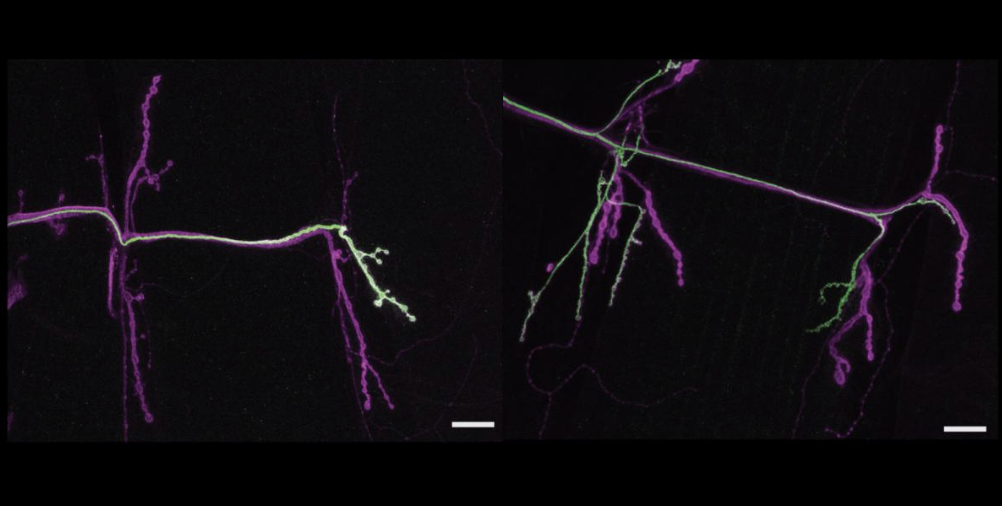
When Weiss joined the Littleton lab after earning her PhD at Hebrew University in Jerusalem in 2013, the lab had just published a new paper about zydeco. It was a long-awaited follow-up. Zydeco disrupted a gene on the fly’s X chromosome which at the time was poorly understood. Led by former graduate student Jan Melom, the lab finally was able to clone the gene that was mutated in Zydeco and showed that it specifically affected “cortex glial” cells and that it caused them to retain too much calcium. But what remained completely unclear was how this made the neurons that those glia contact so susceptible to seizures.
Though Weiss had a research specialty in studying calcium in brain cells, at first she worked on a few other ideas. But because Melom had left the lab, Weiss soon picked up the zydeco baton. In so doing, she was taking on what would become an especially extensive effort involving scores of experiments and a vast array of techniques, some of which she would have to learn along the way.
No hypothesis needed or heeded
Based on the 2013 findings, Littleton had formulated a working hypothesis about what might be going on to cause the seizures. He figured the excess of calcium in the cortex glia probably caused them to emit too much of some kind of signal to the neurons, in turn causing them to remain too active.
That turned out to be wrong, Littleton acknowledges with a smile.
“What Shirley did was to disprove my very strong impression of what was actually happening,” Littleton said. “Sometimes that’s very difficult to do. Once you have an idea of how you think the biology is working, that can reinforce the sorts of experiments you do and affects how you think about the project. It was very exciting in the end that Shirley was able to get past my pre-conceived notions and figure out what was really happening.”
The team didn’t fall into that trap because the experimental approach they chose didn’t depend on what they thought. Weiss’s key initial inquiries were based on a wide-open, free-ranging manipulation of the zydeco flies’ genes. Her strategy was to “knock down” or interfere with the cell’s ability to make use of 847 different genes covering a wide variety of potentially relevant glial cell functions. If knocking down any particular gene stopped the seizures, that would give them a huge clue about how the seizures happen. And whatever worked, if anything, would work regardless of anyone’s guesses up front.
“The great thing about using forward genetics is you don’t have to have a very strong hypothesis,” Weiss said. “You can let the genetics lead the way. I tried to be hypothesis free and to be as unbiased as I could be.”
The knockdown screening yielded about 50 genes where interference totally or partially alleviated the seizures. One in particular squared well with what Melom had observed about a specific cellular process (scientists call it a “pathway”)that related to handling calcium.
Around that time, though, life outside the lab intervened. In February 2015 Weiss and her husband Kfir Sharabi, also a postdoc, and their then four-and-a-half year old daughter, Amit, welcomed their second daughter, Ma’ayan, to the world. With two young kids and the rest of her family in Israel, Weiss came back from maternity leave and got back to investigating the most promising hits of the knockdown screen.
A calcium conundrum
The particular hit related to calcium that caught Weiss’s eye was a gene was called CanB2. Zydeco flies with that gene knocked down experienced no more seizure troubles at all. Moreover, she found that it was specifically helpful to knock it down in cortex glia and that knocking it down in healthy flies didn’t do any discernable harm.
So what does CanB2 do? In general the gene, along with two others, make a protein called calcineurin. No one had ever characterized what calcineurin does in glia. If Weiss could become the first to figure that out, she could whatever problems the zydeco mutation causes.
By manipulating all three calcineurin genes, Weiss was able to confirm that calcineurin activity was indeed crucial for zydeco seizures. She engineered cortex glia so that a glowing green protein would start to be expressed when calcineurin was active. She could see the protein light up under the microscope. This told her there was a lot more calcineurin activity in zydeco mutant brains than in normal fly brains. Apparently, the excess calcium in the cortex glia correlated with increased calcineurin activity.
There are human medicines that ratchet back calcineurin activity. They are typically used to suppress the immune system after a transplant. Weiss wanted to see whether they could reduce seizures in the zydeco flies. When she fed them the drugs the seizures did subside, providing a clear demonstration that intervening in this glial pathway could hold promise for drug development.
Any such effort, to be truly well targeted, would require more than just an association between calcineurin and seizures. Weiss was determined to discover the mechanism that linked the two.
Figuring out what that mechanism was and how it led to seizures, would turn out to be the heart of the discovery and the most challenging phase of her four year endeavor.
A potassium epiphany
Weiss needed to find out what process this excess calcineurin activity might be putting into overdrive. She went back to genetics. She performed a screen to knock down direct targets of calcineurin. It didn’t appear to yield anything helpful. She did another screen of pathways where calcineurin was implicated. In that case, a process called “endocytosis” came up and sure enough, Weiss found that by inhibiting the process in the cortex glia she could again stop seizures in the zydeco flies. Endocytosis is how cells ingest material from their surroundings, including regulating the content of their cell surface membrane proteins. The process can therefore affect the proteins they employ on the membrane to interact with the environment outside the cell. Excess endocytosis could mean that the way by which cortex glia interact with their environment is altered in zydeco, perhaps affecting the neurons they support. But how might that matter in this case?
Weiss struggled with this question for months. She received feedback and advice in meetings with Littleton and in lab meetings where the members discuss, challenge and refine ideas. The discussions were helpful, but it turns out that the key breakthrough came from Weiss attending a conference in Cold Spring Harbor, N.Y. in July, 2018.
Among Weiss’s genetic screen results of calcineurin targets was a gene called “SAND” that makes a protein in flies called “sandman” (the human version is called TRESK). Sandman, when deployed to the cell membrane, forms a channel (picture a portal though the cell’s surface) that allows a cell to bring in potassium ions from outside. At first this result didn’t strike Weiss as all that notable, but at the conference, potassium channels kept coming up as a topic in talks. An idea started to percolate as she took notes. Then at the conference posters she started talking with a scientist who said that problems with potassium channels in glial cells have been linked to epilepsy. Potassium channels apparently merited another look.
“I already had the result,” Weiss said. “I just didn’t connect the two dots.”
One of her screens indeed showed that knocking down SAND in healthy flies caused seizures just like the ones seen in zydeco mutants. Further genetic manipulations confirmed that SAND knockdown and zydeco affected the same pathway in cortex glia cells.
By September 2018, a new hypothesis was emerging: Elevated calcium in cortex glia triggered excess calcineurin activity, which spurred increased endocytosis that hindered sandman’s intake of potassium. This came at a good time as Weiss was nearing the point where she had to start thinking of wrapping up her postdoctoral appointment at MIT. The hypothesis, and the evidence she’d built up, seemed enough to submit a paper to a journal.
Always mindful that a paper was the goal, Weiss had been writing as she went and developing the key figures. When she had a draft done, Littleton then set to polishing it and giving her feedback. eLife wasn’t the first journal they submitted the paper to, but the editors received it enthusiastically. All three of the scientists who reviewed the manuscript for eLife, however, said the same thing: If endocytosis was pulling sandman back from the membrane of the cortex glia, thereby disrupting its ability to take in potassium ions, they wanted to see it happening. Weiss and Littleton not only agreed with that critique, they had even anticipated it.
“You sort of know your own holes in the story,” Littleton said. “This is what we were planning to do next anyway.”
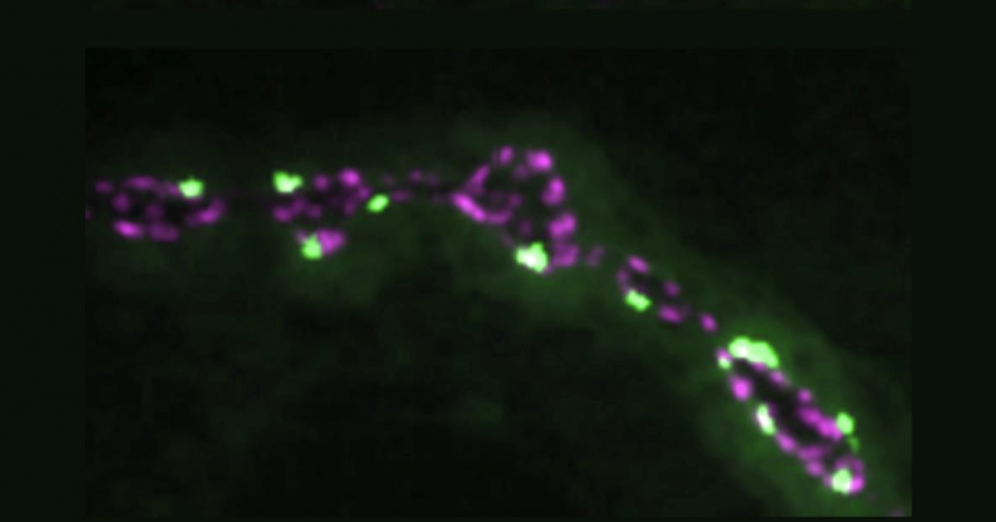
Since sending in the paper, Weiss had already produced those smoking gun images, showing that in zydeco cortex glia, sandman was much less abundant on the membranes than in the non-mutant flies. This cemented the argument, neatly wrapped up in the paper, that neurons become more susceptible to seizing when zydeco cortex glia, saddled with too much calcium and resulting calcineurin activity, overdo the endocytosis of sandman potassium channels, leaving too much potassium outside of neurons, causing increased excitability and the onset of seizures.
Since publication, the paper has garnered some mentions in the scientific press. It has also earned a new National Institutes of Health grant for Littleton’s lab, where they are following in Melom’s and Weiss’ footsteps to study how calcium levels in glia affect the flux of membrane proteins, not just in disease, but as a matter of course in healthy cells. And for Weiss, the paper impressed funders in Israel, providing her with the money to support her new position where she continues studying glia, calcium and seizures.
It was a hard-earned success. Though their end product is knowledge, scientists spend the vast majority of their time with the unknown. Between the lines of most every paper are years of effort in which scientists persistently asked open questions with open minds so that the evidence could lead them to a discovery they could share with the world.