Approach opens the door to a greater understanding of protein-microbe interactions
Lillian Eden | Department of Biology
June 7, 2023
Your mouth is a crucial interface between the outside world and the inside of your body. Everything you breathe, chew or drink interacts with your oral cavity—the proteins and the microbes, including microbes that can harm us. When things go awry, the result can range from the mild, like bad breath, to the serious, like tooth and gum decay to more dire effects in the gut and other parts of the body.
Even though the oral microbiome plays a critical role as a front-line defense for human health and disease, we still know very little about the intricacies of host-microbe interactions in the complex physiological environment of the mouth; a better understanding of those interactions is key to developing treatments for human disease.
In a recent study published in PNAS, a collaborative effort revealed that one of the most abundant proteins found in our saliva binds to the surface of select microbes found in the mouth. The findings shed light on how salivary proteins and mucus play a role in maintaining the oral cavity microbiome.
The collaboration involved members of the Imperiali lab in the Department of Biology and the Kiessling lab in the Department of Chemistry at MIT, as well as the Ruhl group at the University at Buffalo School of Dental Medicine, and the Grimes group at the University of Delaware.
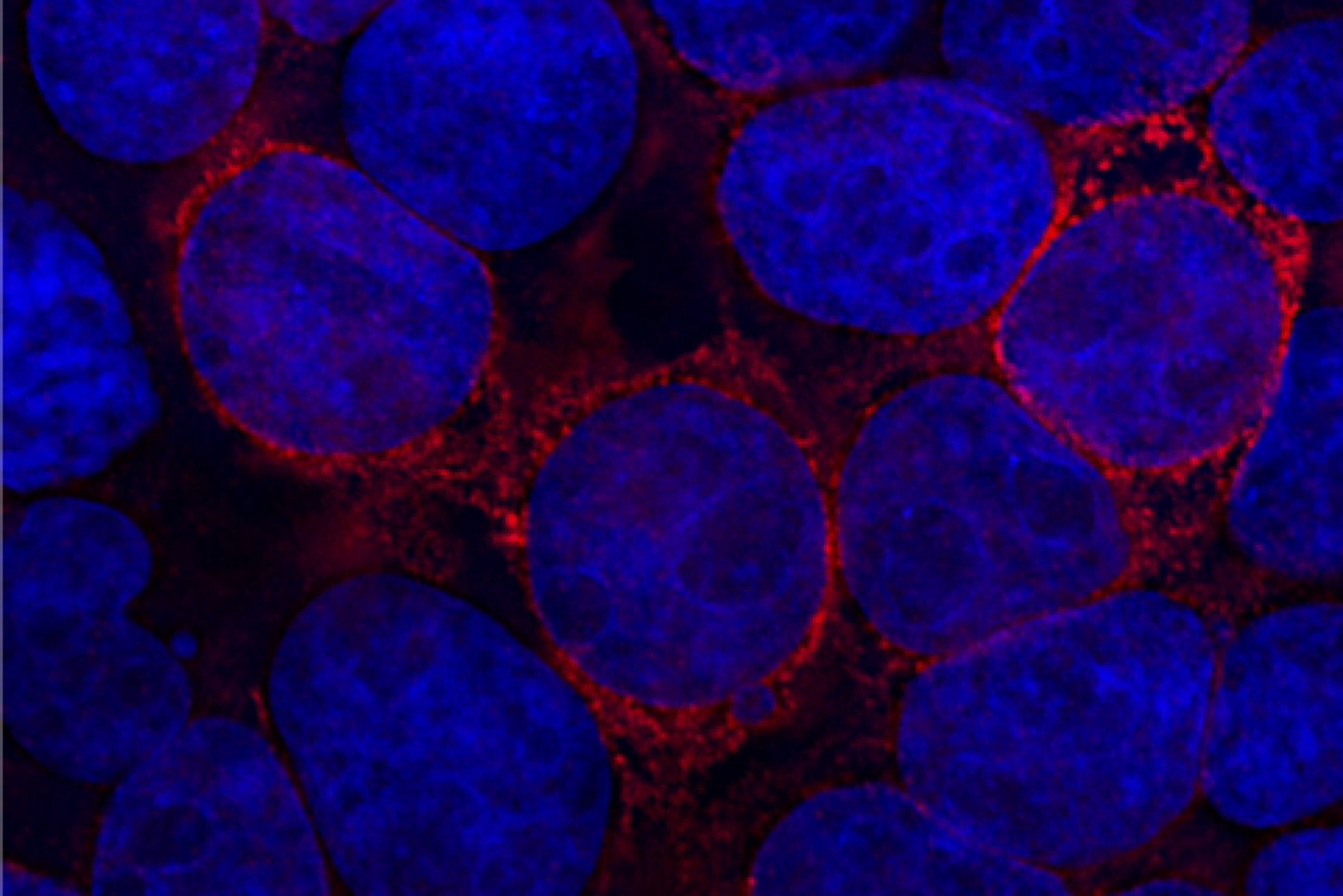
The paper is focused on an abundant oral cavity protein called zymogen granule protein 16 homolog B (ZG16B). Finding ZG16B’s interaction partners and gaining insight into its function were the overarching goals of the project. To accomplish this, Ghosh and colleagues engineered ZG16B to add reporter tags such as fluorophores. They called these modified proteins “microbial glycan analysis probes (mGAPs)” because they allowed them to identify ZG16B binding partners using complementary methods. They applied the probes to samples of healthy oral microbiomes to identify target microbes and binding partners.
The results excited them.
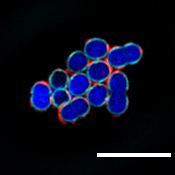
“ZG16B didn’t just bind to random bacteria. It was very focused on certain species including a commensal bacteria called Streptococcus vestibularis,” says first author Soumi Ghosh, a postdoctoral associate in the Imperiali lab.
Commensal bacteria are found in a normal healthy microbiome and do not cause disease.
Using the mGAPs, the team showed that ZG16B binds to cell wall polysaccharides of the bacteria, which indicates that ZG16B is a lectin, a carbohydrate-binding protein. In general, lectins are responsible for cell-cell interactions, signaling pathways, and some innate immune responses against pathogens. “This is the first time that it has been proven experimentally that ZG16B acts as a lectin because it binds to the carbohydrates on the cell surface or cell wall of the bacteria,” Ghosh highlights.
ZG16B was also shown to recruit Mucin 7 (MUC7), a salivary glycoprotein in the oral cavity, and, together the results suggest that ZG16B may help maintain a healthy balance in the oral microbiome by forming a complex with MUC7 and certain bacteria. The results indicate that ZG16B regulates the bacteria’s abundance by preventing overgrowth through agglutination when the bacteria exceed a certain level of growth.
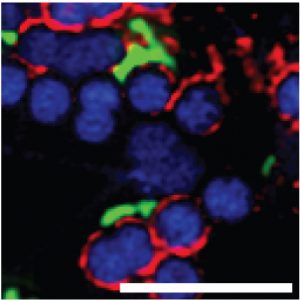
The scale bar shown here represents a 3-micron (µm) length.
“ZG16B, therefore, seems to function as a missing link in the system; it binds to different types of glycans—the microbial glycans and the mucin glycans—and ultimately, maintains a healthy balance in our oral cavity,” Ghosh says.
Further work with this probe and samples of oral microbiome from healthy and diseased subjects could also reveal the lectin’s importance for oral health and disease.
Current attention is focused on developing and applying additional mGAPs based on other human lectins, such as those found in serum, liver, and intestine to reveal their binding specificities and their roles in host-microbe interactions.
“The research carried out in this collaboration exemplifies the kind of synergy that made me excited to move to MIT 5 years ago,” says senior co-author Laura Kiessling. “I’ve been able to work with outstanding scientists who share my interest in the chemistry and the biology of carbohydrates.”
The senior authors of the paper—Barbara Imperiali and Kiessling — came up with the term for the probes they’re creating: “mGAPS to fill in the gaps” in our understanding of the role of lectins in the human microbiome, according to Ghosh.
“If we want to develop therapeutics against bacterial infection, we need a better understanding of host-microbe interactions,” Ghosh says. “The significance of our study is to prove that we can make very good probes for microbial glycans, find out their importance in the frontline defense of the immune system, and, ultimately, come up with a therapeutic approach to disease.”
This research was supported by the National Institute of Health.